原创
摘要:UKMDR下临床研究执行判定
合集:#MHRA
今年七月份,MHRA更新了对CE标识的使用期限。法规指出:根据器械的类型和分类,MHRA将继续接受CE标识器械流通于英国市场直至2030年6月30日。具体时间线如下:
P 符合EU MDD 或EU AIMDD 的一般医疗器械可在英国市场上销售,直至证书到期或2028年6月30日(以较早者为准)为止;
P 符合EU IVDD的IVDs可在英国市场上销售,直至证书到期或2020年6月30日(以较早者为准)为止;
P 在2030年6月30日之前,符合EU MDR的一般医疗器械(包括定制器械)和符合EU IVDR的 IVDs 可在英国市场上销售;
上述时限不适用于欧盟指令规定的I类医疗器械、定制类器械和一般 IVDs。制造商可在2023年6月30日之后自行申报含CE标识的I类器械投放英国市场,前提条件是:
P 根据EU MDR 要求自行申报(直至2030年6月30日);
P 在2021年5月26日之前根据MDD要求进行自我声明,且在MDD下不需要公告机构参与评估,但在EU MDR 下需要公告机构参与评估(直至2028年6月30 日)。这包括升级器械和可重复使用的手术器械。
医疗器械制造商需进行临床研究作为其医疗器械获得 UKCA / CE / CE UKNI 标识过程的一部分,若制造商计划进行医疗器械临床研究,则必须在研究开始前至少60天通知 MHRA。制造商可通过以下流程图判定是否需要提交正式临床研究。
UKMDR2002范围内的研究
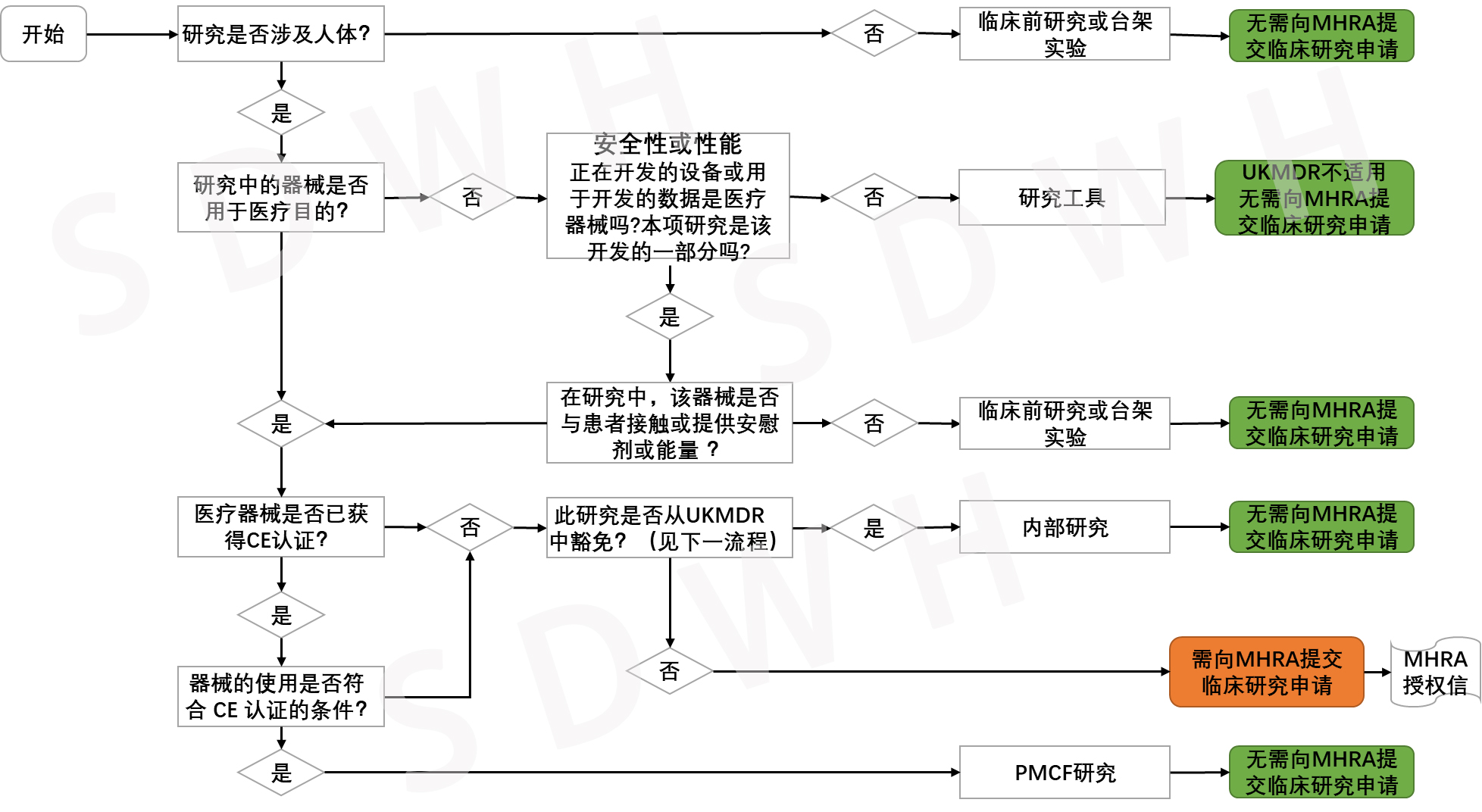
临床前或台架试验:非干预软件研究--利用现有数据提供医疗器械的性能数据,对参与者没有安全影响。可支持符合性评估,但如果不是在人体上进行,则可能不符合临床研究的定义(例如,使用现有患者数据进行软件测试,研究中不与患者互动,研究中不告知患者管理决策)。
医疗机构研究
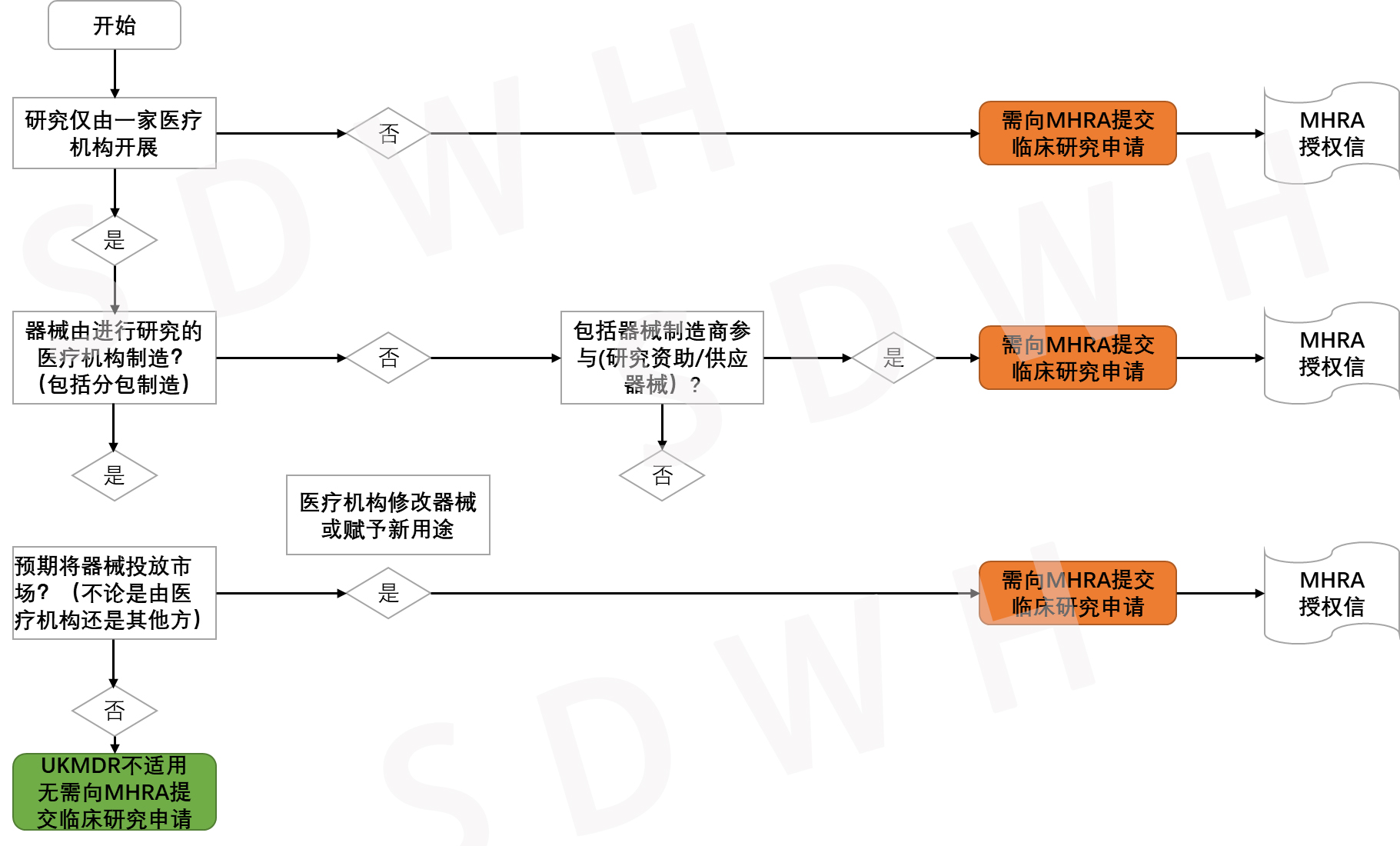
医疗器械制造商/医疗机构提交至MHRA的临床研究申请需阐述以下内容:
若为有医疗用途的临床研究,则应是为符合性评估而执行
若临床研究包括软件研究,其中器械提供的信息将用于为患者管理决策提供依据,或患者在研究中与软件进行互动
若为无医疗用途的临床研究-需提供对参与者有潜在安全影响的性能或安全数据(例如,安慰剂的吸入器/用于健康志愿者的成像系统)
阐明是否将器械转让给第三方(从医疗机构转让给另一家医疗机构,或从制造商转让给医疗机构)
医疗机构需澄清是否有意将器械投放市场
注:若器械用于医疗目的,但其临床研究可能不支持符合性评估,而是包括研究治疗或以研究为目的的研究。(例如
深部脑刺激器旨在研究脑电波,但在研究期间也用于提供治疗)。则这类研究将需要修改,以涵盖安全/性能目标)。
参考资料:






