原创
摘要:MDCG发布MDR对医疗器械制造商的语言要求
合集:#欧盟MDR法规合集
MDR包含不同的法律规定,允许成员国在国家层面确定对制造商随附器械信息的语言要求。下表概括介绍了欧盟成员国在确定制造商语言要求方面的国家规定。虽然欧盟成员国没有义务确定具体语言,但考虑到以各种语言提供信息的相关费用,欧盟主管当局鼓励各成员国考虑如果不影响器械的安全使用,尤其是需要专业人士使用的器械,是否可以接受制造商以本国语言以外的另一种语言(如英语)提供信息.
关于下表欧盟各成员国语言要求的相关法律条款,感兴趣的可戳链接:md_sector_lang-req-table-mdr.pdf (europa.eu)
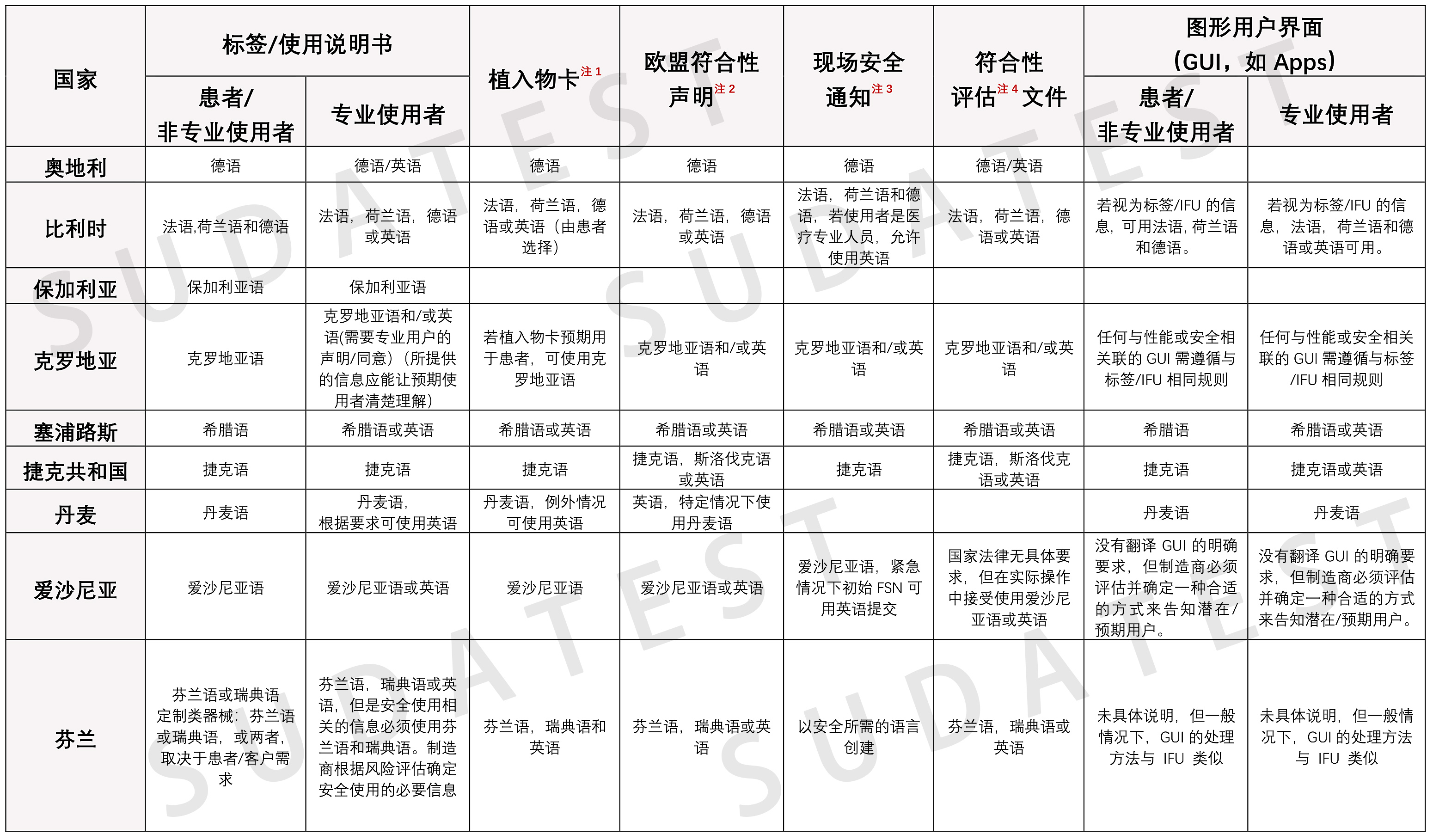
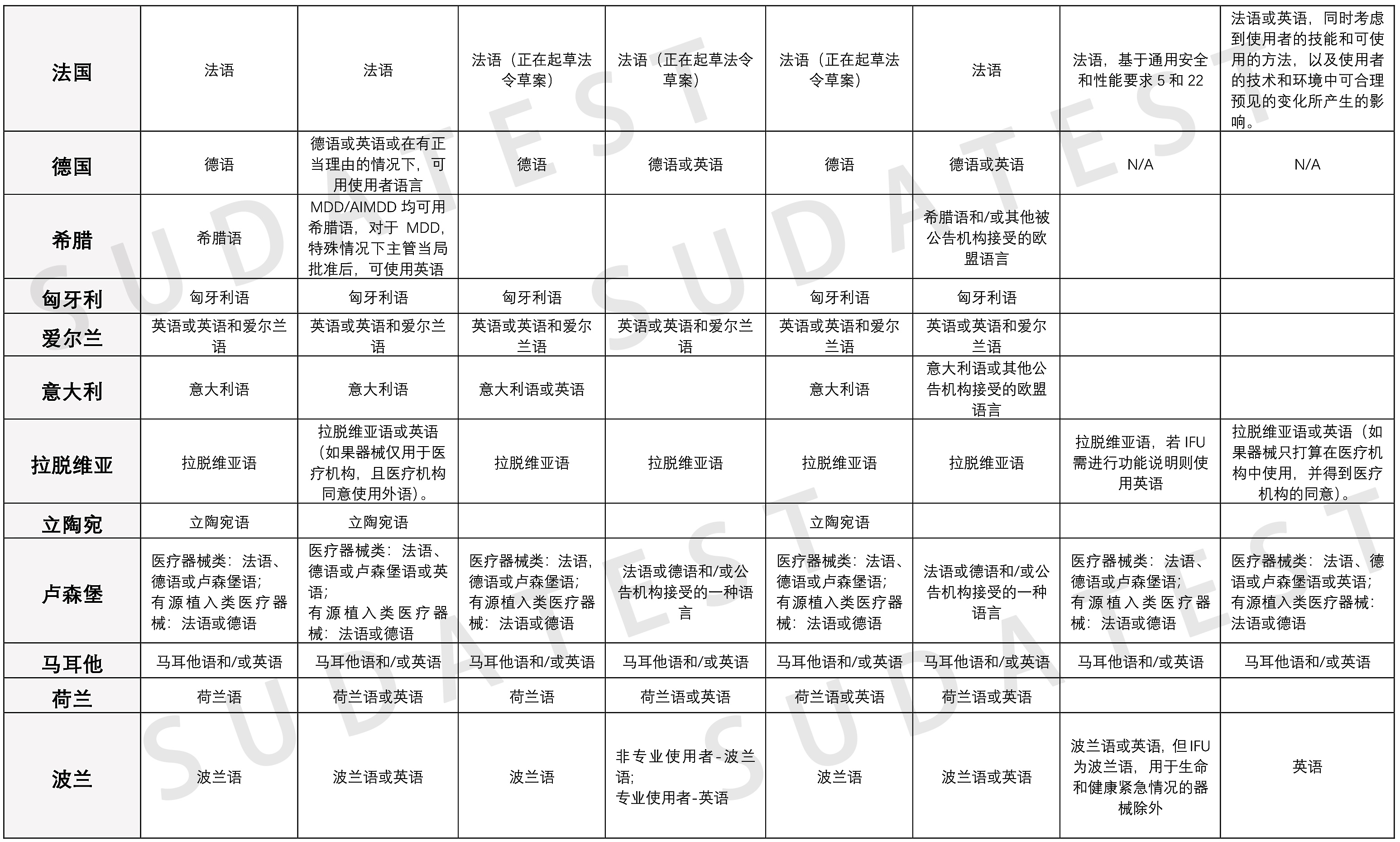
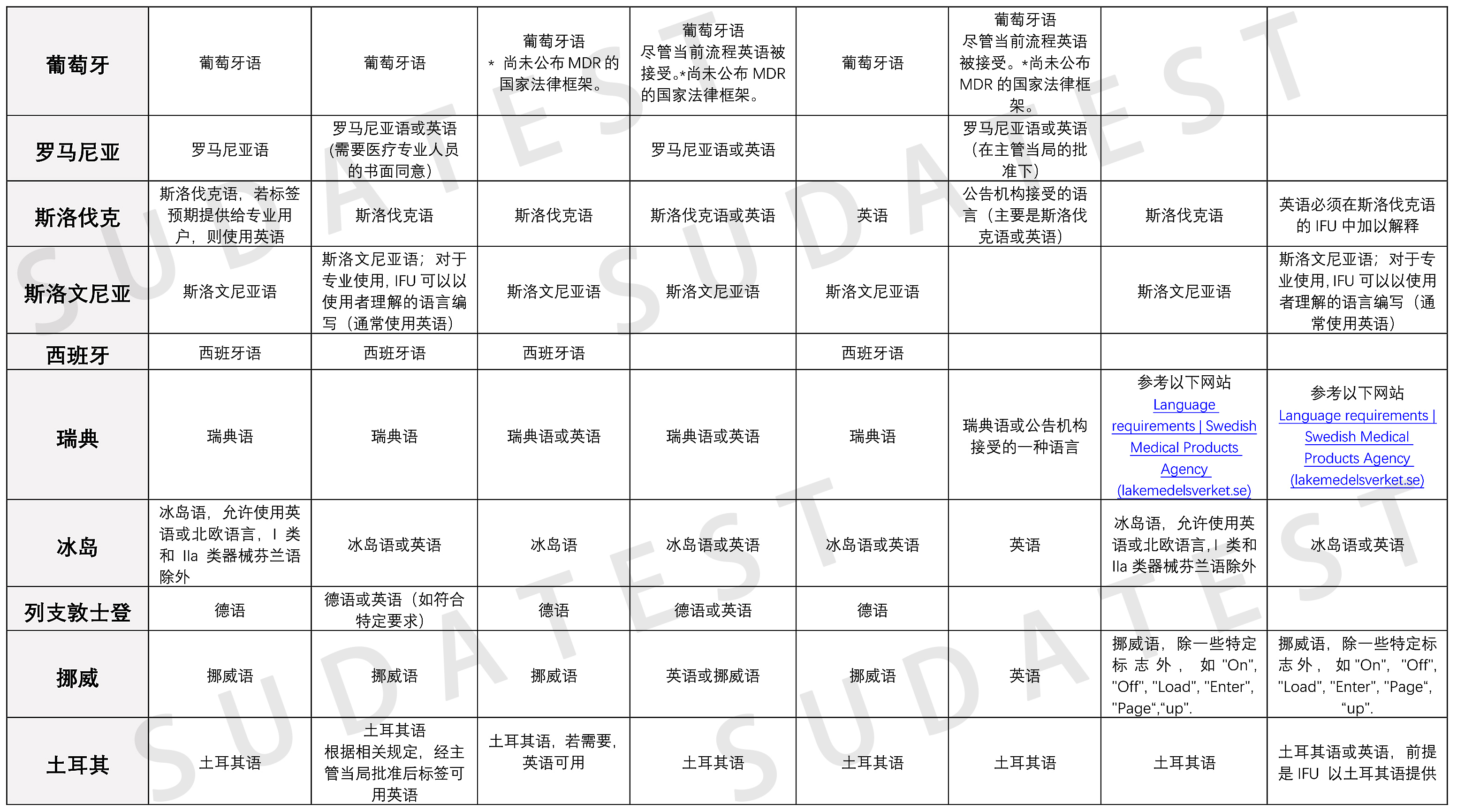
注1:植入物卡:根据MDR Art.18规定,制造商应在与器械一起交付的植入物卡中提供器械标识信息,包括器械名称、序列号、批号、UDI、器械型号,以及制造商名称、地址和网站。
注2:欧盟符合性声明:根据MDR Art. 19规定,欧盟符合性声明须说明已履行MDR中相关涵盖器械规定的要求。制造商应当不断更新欧盟符合性声明。欧盟符合性声明至少应包含列于MDR 附录IV中的信息,且应将其翻译成欧盟官方语言或者器械销售所在成员国所要求的语言。
注3:现场安全通知:根据MDR Art. 2 (69)指制造商向使用者或客户发送的与现场安全性纠正措施相关的通知。
注4:符合性评估:根据MDR Art.2(40)规定,指证明MDR中与器械相关的要求是否得到满足的过程。
参考资料:
【供 稿】苏大检测医疗器械事业部法规中心






