原创
摘要:医疗器械再制造最终指南发布,明确了维修和再制造的区别,规定了再制造的具体活动表现以及再制造企业的监管要求。
合集:#FDA
简介
医疗器械包含大量产品,其技术、生命周期、复杂程度、预期用户和使用环境各不相同。许多器械都是可重复使用的,在其使用寿命期间需要进行预防性维护和修理。然而,器械的 "维修"和 "再制造"之间的区别并不明晰,因此FDA一直致力于明确“维修”和“再制造”之间的差别。2024年5月10日FDA正式发布“医疗器械再制造”最终指南,旨在帮助澄清在器械上进行的活动是否可能是"再制造"。
范围
本指南针对的是在打算重复使用和维护的器械上进行的活动,讨论原始设备制造商(original equipment manufacturer, OEM)和第三方对此类器械进行的活动是否可能属于再制造。本指南范围内的产品适用于所有I、II、III类可重复使用的器械,包括符合器械定义的软件和电子产品;但不涉及再加工的一次性使用器械。
“再制造”和“维修”的含义
再制造:指对成品器械进行加工、调节、翻新、重新包装、复原或任何其他行为,从而显著改变成品器械的性能、安全规格或预期用途。本指南中,FDA 将OEM 合法销售的成品器械称为 "已合法上市器械(legally marketed device)"。
维修:指在销售后对成品器械中的一个或多个部件进行修理和/或预防性或例行维护,以使其恢复到OEM规定的安全和性能规格,并满足其最初的预期用途。
确定“再制造”活动指导原则
FDA建议使用以下指导原则确定活动是否属于“再制造”:
P 评估在预期用途上是否有变化。
P 确定活动单独或累计是否会显著改变成品器械的安全或性能规格。
P 评估器械的任何变更是否需要提交新的上市申请。
P 评估组件/部件/材料的尺寸和性能规格,可通过与OEM的组件/部件/材料规格进行比较和/或通过验证和确认测试进行评估。
P 基于ISO 14971开展基于风险的方法进行评估。当基于风险的评估发现任何新的风险或对已知风险的重大修改时,该活动就可能是再制造。因为与已合法上市器械相比,这些风险或修改可能会显著改变性能或安全规格。
P 充分记录决策过程。充分的记录有助于再制造商对不同时期同一类型的活动进行认定决策,并在FDA进行检查或以其他方式要求提供信息时,帮助证明其决策的合理性。
确认活动是否是“再制造”的相关考虑
对于器械而言,FDA已确定了某些类型的活动会严重改变已合法上市器械的性能和安全规格。如器械灭菌方法的变更、器械再处理说明的变更以及器械控制机制、操作原理或能量类型的变更。
对于包含组件/部件/材料的活动,FDA建议使用以下流程图进行判定其活动是否为“再制造”。
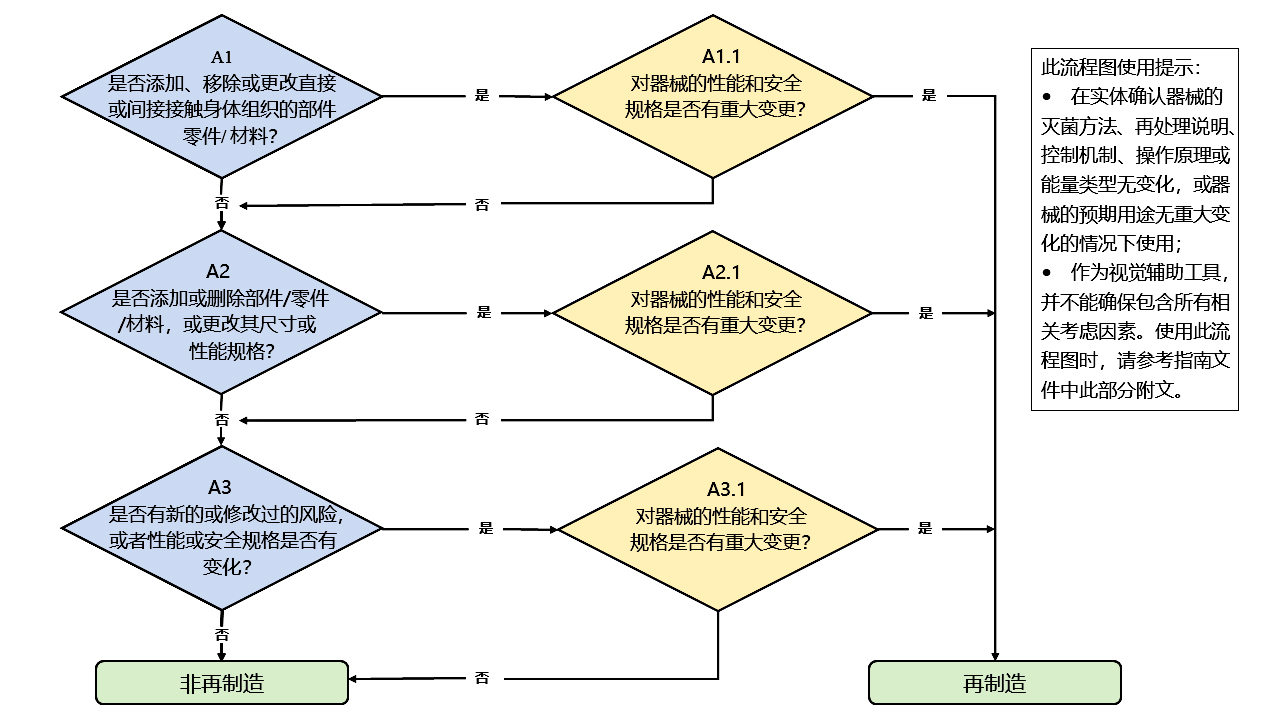
图1. 判定执行的活动是否属于再制造的流程图
软件变更的判断
通常软件的变更会引起其架构、要求规格、未解决的异常情况和其他关键特征的变化,因此许多软件变更很可能是再制造。此外,由于软件故障的概率无法用传统的统计方法确定,因此FDA在上述文章中建议的流程图判定法以及基于风险的评估方法均不适用于软件变更。相反,FDA已确定对软件进行的某些活动可能不属于再制造:
P 代表OEM或经OEM明确授权,使已合法上市器械恢复到其性能和安全规格,或保持其性能和安全规格以及预期用途的活动;
P 实施OEM授权、批准或以其他方式提供的更新和升级;
P 运行基于软件的硬件诊断;
P 评估病毒、恶意软件和其他网络安全相关问题;
应注意:判断软件变更是否属于再制造也应考虑其累积影响。
再制造企业的监管要求和注意事项
根据 FD&C Act 和 21 CFR Part 820的规定,再制造者被视为制造商,通常须遵守与OEM相同的监管要求,包括但不限于:
P 工厂注册和器械列示;
P 上市许可;
P 医疗器械不良事件上报和电子产品报告;
P 纠正和删除报告及缺陷通知;
P 质量体系符合性;
P 标签……
标签的考虑
若再制造的器械明显偏离OEM的标签,则再制造商需要进行相应的标签更改申请,使其恢复到OEM所规定的性能和安全规格。为促进和保障公众健康,FDA鼓励OEM作为行业最佳实践,向再制造商提供至少包括以下信息的OEM标签,以便于对可重复使用的器械进行日常维护和修理:
关键性能和安全规格的描述;
关键技术或功能规格,包括物理尺寸、电气特性、内部保险丝和电源的说明;
器械特定的性能规格;
建议的维修活动和时间表;
建议的故障排除步骤、例行测试和验收标准,以确认器械仍符合其性能和安全规格;
器械错误代码、警报和报警功能说明;
与器械维修相关的注意事项和警告;
软件版本号和发布日期。
【供 稿】苏大检测医疗器械事业部法规中心






