上周我们介绍了MDR全流程认证共识文件的目的和范围,认证流程各阶段中的首次联系和预申请提交、预申请评审和报价、递交正式申请、合同/书面协议和申请评审四个环节。(回顾请戳链接:【MDR】认证企业必读,Team-NB发布MDR全流程认证的共识文件(上))本文我们将继续介绍其他四个环节以及语言要求。
符合性评估
在接受正式申请并签订书面协议后,NB将对申请进行审查,并制定计划,为每个项目开展适当的符合性评估活动,包括在适用的情况下开展物理测试、实验室测试或其他测试。符合性评估活动的选择取决于器械的分类和所选择的符合性评估程序。NB会告知制造商计划开展所需的符合性评估活动的期限。各种器械分类所需的典型符合性评估活动如下:
I类器械:其中Im、Is、Ir类器械均须进行 NB 符合性评估。不过,对于这些类型的器械,NB的干预仅限于:Im类器械计量方面、Is类器械建立、确保和维护无菌屏障有关的方面,以及Ir类器械重复使用有关的方面(清洁、消毒、灭菌、维护和功能测试以及相关使用说明)。
IIa、IIb和III类器械:需结合QMS审核、技术文件审核以及基于所选符合性途径的器械测试。除这些活动外,根据器械的性质,可能还需要执行其他特定的附加程序/流程。
对于QMS评估,审核在制造商的厂房内进行,必要时也会在制造商的供应商和分包商的厂房内进行。NB根据其审核规则和程序确定制造商的QMS是否符合法规要求。如果申请中包含的一种或多种器械是无菌的,一些NB还可能会进行单独的微生物审核。NB在审核结束后会发布一份QMS审核报告,记录审核结果,包括根据审核结果提出认证建议(或拒绝认证)。如果任何审核结果被定性为重大不符合项,制造商须及时全面地去解决这些问题,并由NB在额外的审核中进行核实后才能提出认证建议。
对于技术文件评估,应符合MDR Annex II和Annex III中规定的要求,具体如下:
- 根据MDR Art.52(4)的规定,系统地评估每种III 类/IIb 类植入器械(属于成熟技术(WET)的器械除外,如针、螺钉等);
-对 IIb 类 WET/非植入性和 IIa 类医疗器械遵循MDCG 2019-13进行抽样。
完成技术文件审核后,NB将发布技术文件评估报告(TDAR)和临床评价评估报告(CEAR),记录评估结果、结论以及根据结论提出的认证(或拒绝认证)建议。对于NB开出的不符合项,制造商必须提供CAPA,并尽职尽责地处理不符合项。根据发现问题的性质、严重程度和复杂性以及需要采取的行动,在NB提出认证建议之前,可能需要进行额外的审核/评估。如果选择的符合性评估途径包括MDR Annex X(型式检验)或Annex XI Part B(产品验证),那么NB将根据适用附件的要求对器械进行额外测试。
特定程序:除上述QMS审核、技术文件评估和测试外,根据器械的分类和其他功能/特征,还可能适用下表中一种或多种程序。
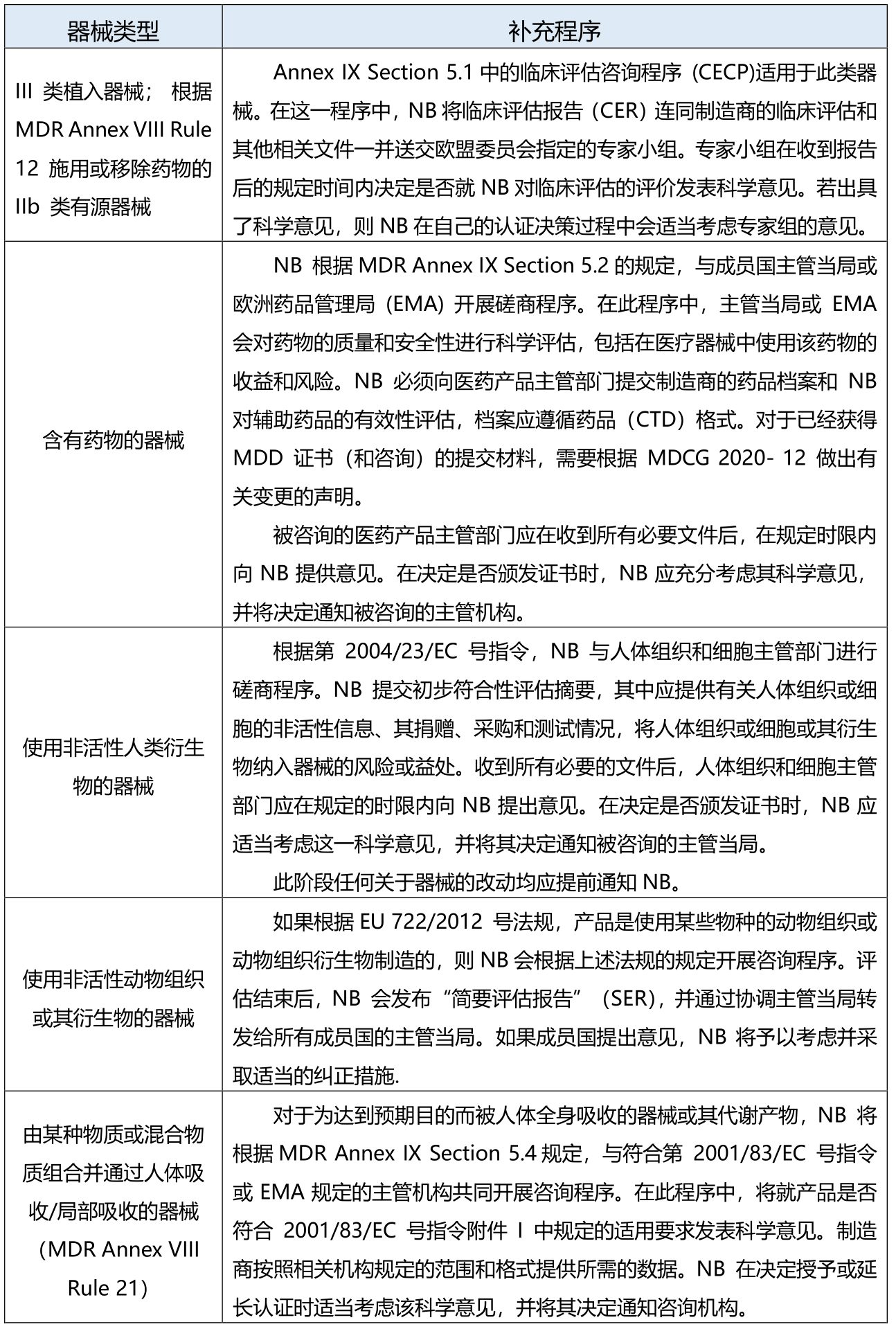
根据MDR Art.22(3)的系统和程序包: NB的参与仅限于确保与无菌有关的程序方面,直至无菌包装被打开或损坏。
III类定制植入器械:NB的参与仅限于评估制造商为遵守适用要求而建立的QMS。
最终评审和做出决定
一旦完成所需的符合性评估活动,NB将进行最后的审查和决策步骤,根据评估活动的结果和建议颁发证书或拒绝颁发证书。最终评审由未参与过申请器械符合性评估程序的人员进行。最终评审应核实:
- 用于决策的报告和证明文件,包括有关解决评估过程中注意到的不符合项的报告和证明文件,在申请范围方面是完整和充分的,
- 不存在妨碍颁发证书的未解决不符合项。
审查的有利或不利结果通常反映在一份内部报告中,该报告作为一份摘要,包含认证过程的主要阶段、评估结果,并在最后就是否颁发证书提出建议,作为NB相关人员决策过程的一部分。决策过程会考虑到最后审查步骤的建议、评估文件和其他相关补充信息,以决定是否满足了MDR的要求。除其他事项外,决策步骤还应考虑上市后监管计划(包括 PMCF 计划)的适当性、需要为NB进一步审查最新临床评估设定的任何具体里程碑、需要为认证定义的任何具体条件或规定,以及不超过五年的认证期限。
签发证书
如果决策过程最终决定签发证书,则NB将根据适用的符合性途径生成证书,其中包含 MDR Annex XII 中规定的信息。证书将发放给制造商,并提交至EUDAMED(根据EU 2023/1860 号文件规定的时限必须提交)。
上市后监管
在初始认证结束时NB会确定维持所颁发证书所需的监管活动。NB 还将不断更新监管计划,包括对合法制造商及其分包商/供应商(如果相关)进行年度 QMS 审核、PSUR 评估、SSCP 验证、技术文件抽样评估、警戒数据评估、突击审核等。
制造商必须制定一套程序,将其QMS或器械的任何重大变更计划通知NB。通知要求基于所遵循的符合性评估途径。NB必须评估所提出的变更,并核实在这些变更之后,QMS或器械的设计/类型是否仍然符合MDR的要求,并将其决定通知制造商。根据变更的性质,NB可能需要开展额外的符合性评估活动,如QMS审核或技术文件评估,以支持变更的批准。
语言
技术文档和QMS文档的语言要求由NB具体规定。






