2025年1月13日,美国FDA发布了《上市前批准申请(PMA)和人道主义器械豁免(HDE)模块审查》的最新指南。
背景介绍
传统的PMA/HDE申请中,申请人需同时提交所有数据,且FDA仅在收到所有所需信息后才开始审查。1998年,FDA引入一种替代性监管途径,允许采用模块化方法提交PMA申请。2003年10月26日,《2002年医疗器械用户费用和现代化法案》(MDUFMA)正式生效,以法律形式确立了模块化审查方法,至此,模块化审查方法也适用于HDE申请。
模块化审查方法旨在为申请人提供一种机制,使其在准备申报器械临床数据的同时,提交非临床数据和生产信息供FDA审查。因此,模块化的PMA/HDE是由在不同时间提交的“模块”组成,这些模块组合在一起成为一个完整的申请。模块化审查方法允许申请人在完成非临床测试和分析之后尽快将其他模块提交给FDA进行审查,从而提高审查过程的效率。此外,模块化审查方法还允许申请人在审查过程比传统PMA/HDE申请更早地解决FDA提出的任何缺陷。
范围
模块化审查方法是传统PMA/ HDE准备、提交和评估的一种替代方式,适用于处于临床研究早期阶段的产品。当申请人接近提交原始PMA/HDE,或器械设计不稳定或可能发生变化时,模块化审查方法并不适用。
由于FDA认为PMA/HDE的补充申请很少适合进行模块化审查,因此本指南的范围仅限于申请原始PMA/HDE批准的申请人。
提交模块化PMA/HDE流程
联系审核组
第一步应先通过电子邮件联系CDRH/CBER相关审查部门的助理主任/部门主任,表明提交模块化PMA/HDE的意向,具体流程如下:
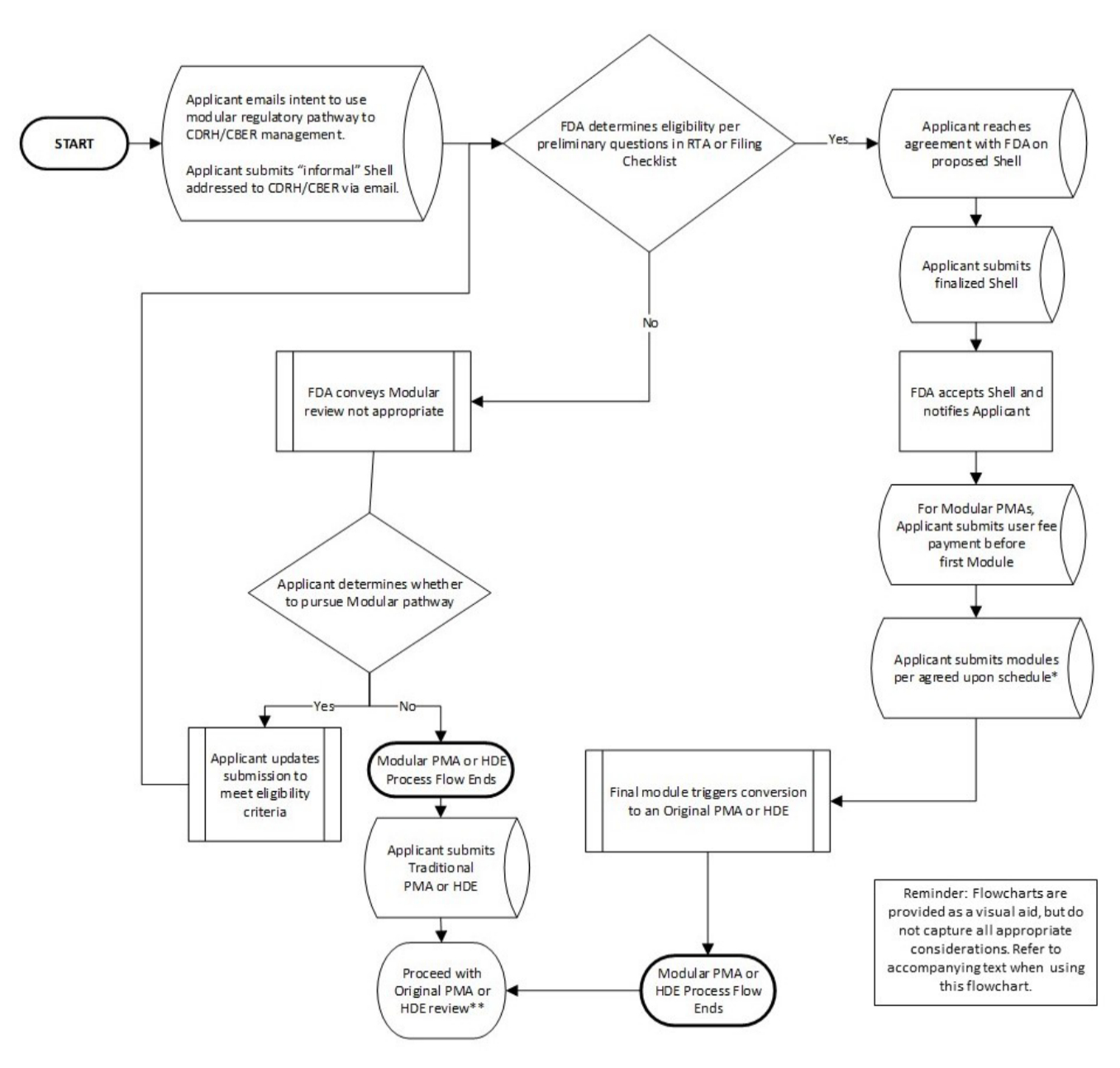
PMA/HDE拟议框架内容准备及申请非正式审查
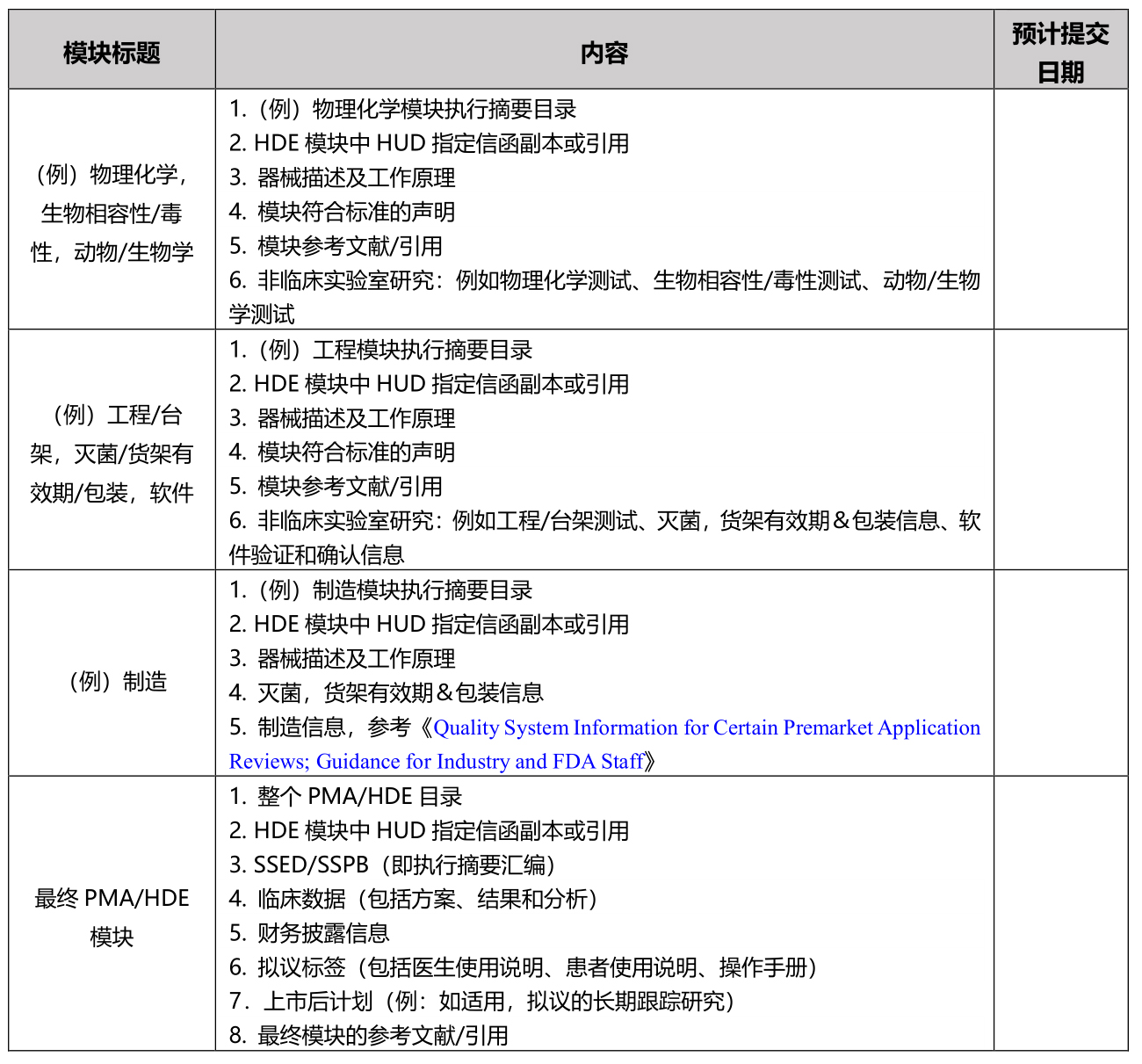
申请人通过电子邮件联系相关人员的同时,应附上拟议框架内容。每个模块的框架内容应进行详细描述,以便FDA能够全面了解计划提交的各个模块。可参考下表进行框架内容设计。
公司/器械名称
申请人不应计划连续提交模块,模块的提交时间表应允许审查部门在收到下一个模块之前完成对本模块的审查(至少90天)。如果资源允许,在与审查组充分讨论后,制造模块可与其他模块同时进行审查。
在正式提交到文件控制中心(DCC)之前,拟议的模块化框架内容应与FDA审查团队进行互动讨论并达成一致。非正式审查的步骤是1)确认PMA/HDE审查是否适用于该申报器械,2)审查拟议框架内容。
正式提交最终框架内容
在与FDA就拟议框架内容达成一致后,可进行最终框架内容的提交。若是提交至CDRH的,则通过“CDRH门户网站“以电子方式提交最终框架内容,或通过邮寄方式;若是提交至CBER的,则通过FDA电子提交网关以电子方式提交,或通过CBERDCC_eMailSub@fda.hhs.gov 提交不超过150MB的文件,或通过邮寄方式。
对已接受框架内容的变更
如果在正式提交并接受框架内容后需要进行变更(例如,器械设计变更导致需要额外或不同的测试,或者模块数量增加或减少),申请人应通过电子邮件通知审查团队,并说明变更的必要性。FDA在非正式审查这些变更后,将会与申请人互动讨论。在获得审查团队同意后,可对模块的提交时间和顺序进行调整。对于模块内容、标题或数量的重大变更,应正式提交修订后的框架,如同提交最终框架一样。
PMA/HDE模块
每个模块的提交
申请人应将提交内容明确标识为模块化提交,并在封面信中引用之前分配的框架内容编号和模块标题。FDA将按照收到的模块顺序依次分配模块编号。模块包含的内容参考上表,模块之间重复一些信息(例如器械描述)是必要的,以便分配到该模块的审查员能够高效地进行审查。
模块审查的时间框架
FDA的目标是在收到模块后的90天内完成每个模块的审查,并发出缺陷信或接受通知。模块化审查本质是在合理范围内进行互动。每个模块可以在框架内容中确定的预计提交日期内继续提交,无论之前模块的审查状态如何。任何模块的预计提交日期的变更都应通过电子邮件与FDA审查团队沟通,并达成一致。
如果模块提交不完整,FDA和申请人将无法充分享受到模块化审查的好处。因此,申请人应完成所有支持该模块所需的测试。如果提交的模块不完整,FDA计划向申请人发出缺陷信,指出审查无法继续进行,直到收到缺失的信息。FDA计划在收到包含缺失信息修正后的90天内完成对该模块的审查。
如果在FDA判定模块可接受之后对器械进行了修改,则申请人应联系相应的审查人员讨论并进行非正式审查以评估该修改以及支持该修改所需的任何测试。如果审查人员认为该修改是一项重大变更,申请人应向每个受影响的模块提交修正,其中应包含修正后的执行摘要、变更描述、重复进行的测试清单以及为支持该修正而生成的数据,且这一修正将重新开启相关模块。若申请人提交模块之后,适用的政策和法规发生了变化,FDA建议联系审查人员讨论是否需要更新已提交的模块,以及如何提交这些更新,未作为修正提交的更新应包含在最终模块中。
申请人提交最终模块(即最终临床数据、拟议标签及PMA的SSED或HDE的SSPB),并引用之前提交的模块,完成模块化申请。申请人应在封面信中明确标识最终模块提交为“已完成的模块化PMA/HDE”,并具体引用已被FDA接受的模块(通过框架和模块标题),并指出任何存在未决缺陷的模块。为了使PMA/HDE申请完整,申请人应在最终模块中提供对所有未决缺陷的回复。
在收到最终模块后,FDA将把已完成的模块化申请转换为原始PMA/HDE,并为其分配PMA/HDE编号。
完成模块化申请转换后,审查将按照原始PMA/HDE的流程进行,FDA根据最终模块是否包含完成申请所需的所有信息来做出接受和/或备案的决定。具体要求如下:
PMA: FDA计划使用指南《Acceptance and Filing Reviews for Premarket Approval Application (PMAs)》中描述的信息做出接受和备案的决定。与传统PMA类似,FDA通常会在收到最终模块后的15天内做出接受或拒绝接受的决定。如果接受,则在收到最终模块后的45天内做出备案或不备案的决定。如果FDA决定备案PMA,备案日期为申请完成的日期,通常是最终模块的接收日期。PMA的180天审查周期也将从该日期开始。若FDA决定不接受PMA,则发出通知,指出缺失项;若FDA决定不备案PMA,则向PMA申请人发出不备案决定。
HDE:FDA计划使用指南《Humanitarian Device Exemption (HDE) Program》中描述的信息要求备案HDE之前需要提交的信息。FDA将在收到最终模块后的30天内做出备案或不备案的决定。若FDA决定备案,备案日期为申请完成的日期,通常是最终模块的接收日期,HDE的75天审查周期也将从该日期开始;若FDA决定不备案,则向HDE申请人发出不备案决定。
参考资料:Premarket Approval Application and Humanitarian Device Exemption Modular Review






