【声明】版权所有,未经许可不得转载使用!
近日,国家市场监管总局发布修订后的《医疗器械生产监督管理办法》,并自2022年5月1日起施行。
总则
从事医疗器械生产活动,应当遵守法律、法规、规章、强制性标准和医疗器械生产质量管理规范,保证医疗器械生产全过程信息真实、准确、完整和可追溯。
医疗器械注册人、备案人对上市医疗器械的安全、有效负责。
根据医疗器械风险程度,医疗器械生产实施分类管理。
生产许可管理
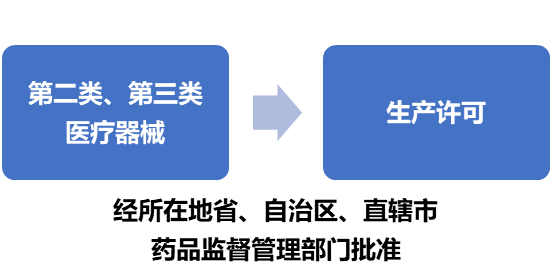
在境内从事第二类、第三类医疗器械生产的,应当向所在地省、自治区、直辖市药品监督管理部门申请生产许可,并提交下列材料:
(一)所生产的医疗器械注册证以及产品技术要求复印件;
(二)法定代表人(企业负责人)身份证明复印件;
(三)生产、质量和技术负责人的身份、学历、职称相关材料复印件;
(四)生产管理、质量检验岗位从业人员学历、职称一览表;
(五)生产场地的相关文件复印件,有特殊生产环境要求的,还应当提交设施、环境的相关文件复印件;
(六)主要生产设备和检验设备目录;
(七)质量手册和程序文件目录;
(八)生产工艺流程图;
(九)证明售后服务能力的相关材料;
(十)经办人的授权文件。
Note
申请人应当确保所提交的材料合法、真实、准确、完整和可追溯。
相关材料可以通过联网核查的,无需申请人提供。
省、自治区、直辖市药品监督管理部门应当对申请资料进行审核,按照国家药品监督管理局制定的医疗器械生产质量管理规范的要求进行核查,并自受理申请之日起20个工作日内作出决定。现场核查可以与产品注册体系核查相结合,避免重复核查。
符合规定条件的,依法作出准予许可的书面决定,并于10个工作日内发给《医疗器械生产许可证》。

生产备案管理
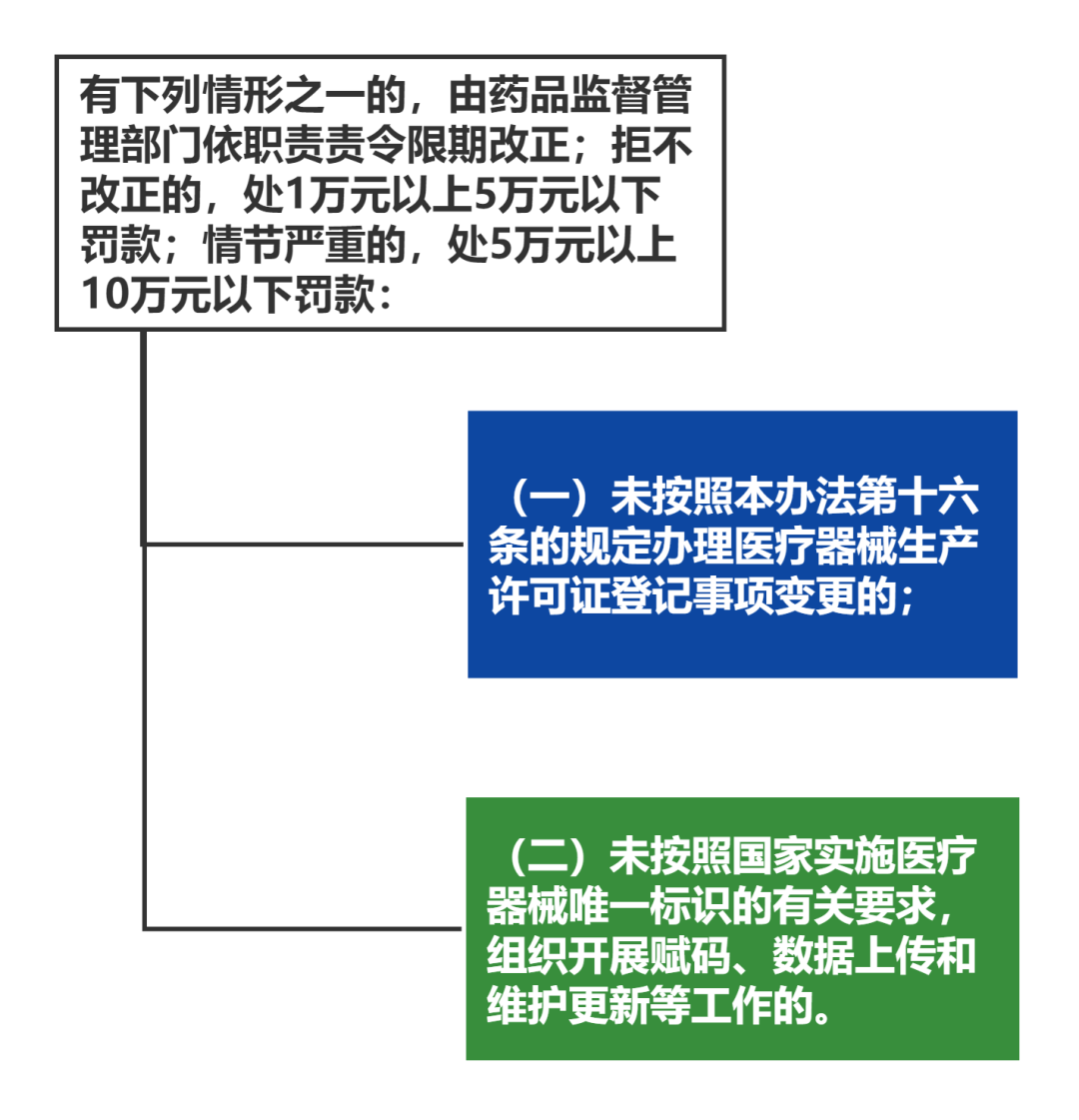
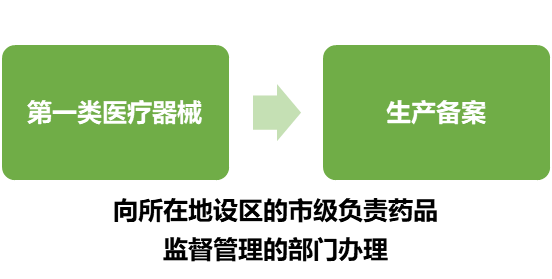
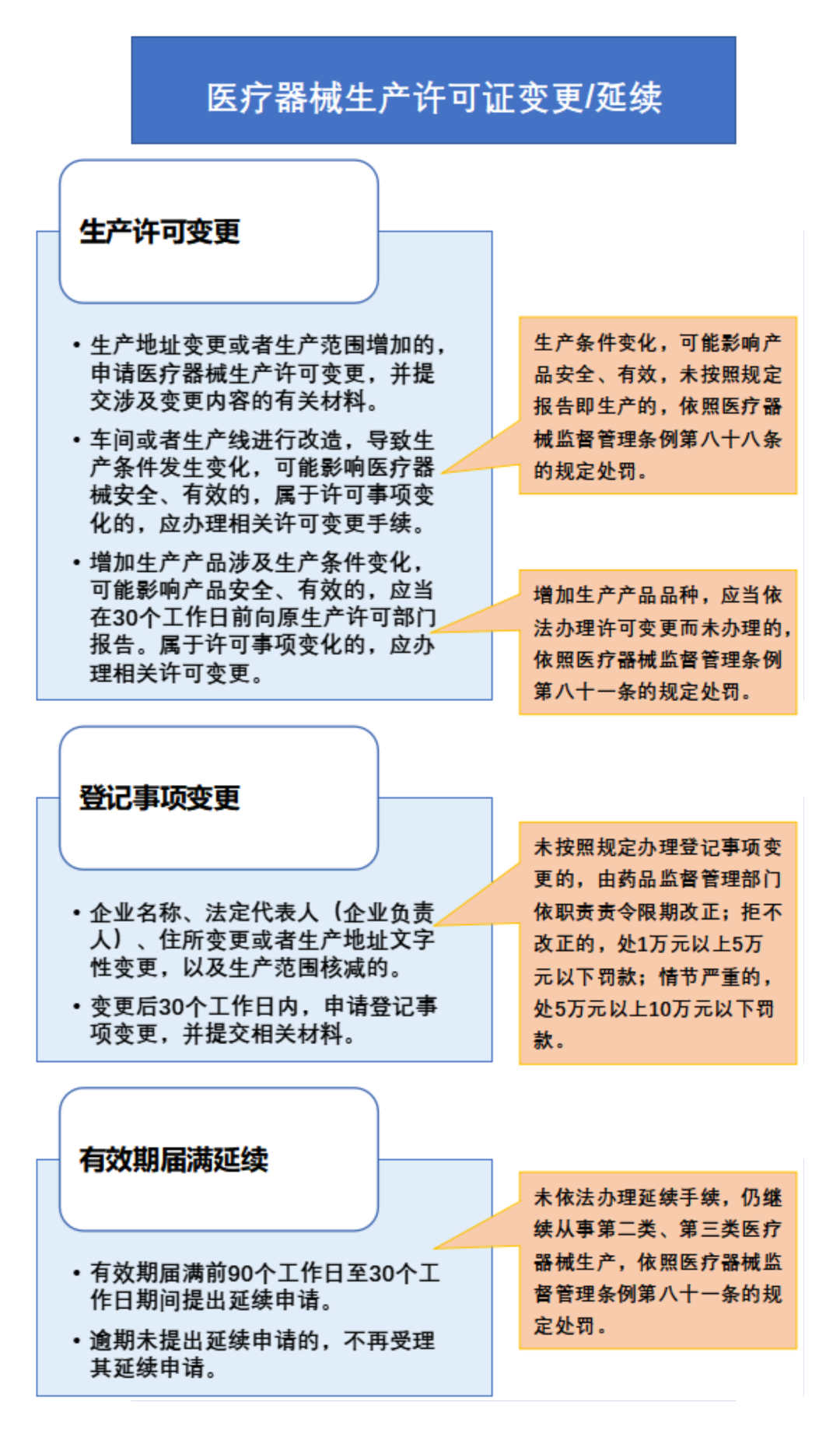
从事第一类医疗器械生产的,应当向所在地设区的市级负责药品监督管理的部门备案。医疗器械备案人自行生产第一类医疗器械的,可以在办理产品备案时一并办理生产备案。第一类医疗器械生产备案内容发生变化的,应当在10个工作日内向原备案部门提交与变化有关的材料。
增加生产产品品种的,应当向原生产备案部门报告,涉及委托生产的,还应当提供委托方、受托生产产品、受托期限等信息。
未按照本办法规定办理第一类医疗器械生产备案变更的,依照医疗器械监督管理条例第八十四条的规定处理。
生产质量管理
医疗器械注册人、备案人、受托生产企业应当按照医疗器械生产质量管理规范的要求,建立健全与所生产医疗器械相适应的质量管理体系并保持其有效运行,并严格按照经注册或者备案的产品技术要求组织生产,保证出厂的医疗器械符合强制性标准以及经注册或者备案的产品技术要求。
Note
违反医疗器械生产质量管理规范,未建立质量管理体系并保持有效运行的,由药品监督管理部门依职责责令限期改正;影响医疗器械产品安全、有效的,依照医疗器械监督管理条例第八十六条的规定处罚。
医疗器械注册人、备案人的法定代表人、主要负责人对其生产的医疗器械质量安全全面负责。
医疗器械注册人、备案人、受托生产企业应当配备管理者代表。管理者代表受法定代表人或者主要负责人委派,履行建立、实施并保持质量管理体系有效运行等责任。
医疗器械注册人、备案人委托生产的,应当对受托方的质量保证能力和风险管理能力进行评估,按照国家药品监督管理局制定的委托生产质量协议指南要求,与其签订质量协议以及委托协议,监督受托方履行有关协议约定的义务。
医疗器械注册人、备案人应当负责产品上市放行,建立产品上市放行规程。委托生产的,医疗器械注册人、备案人还应当对受托生产企业的生产放行文件进行审核。
医疗器械注册人、备案人应当建立并实施产品追溯制度,保证产品可追溯。受托生产企业应当协助注册人、备案人实施产品追溯。
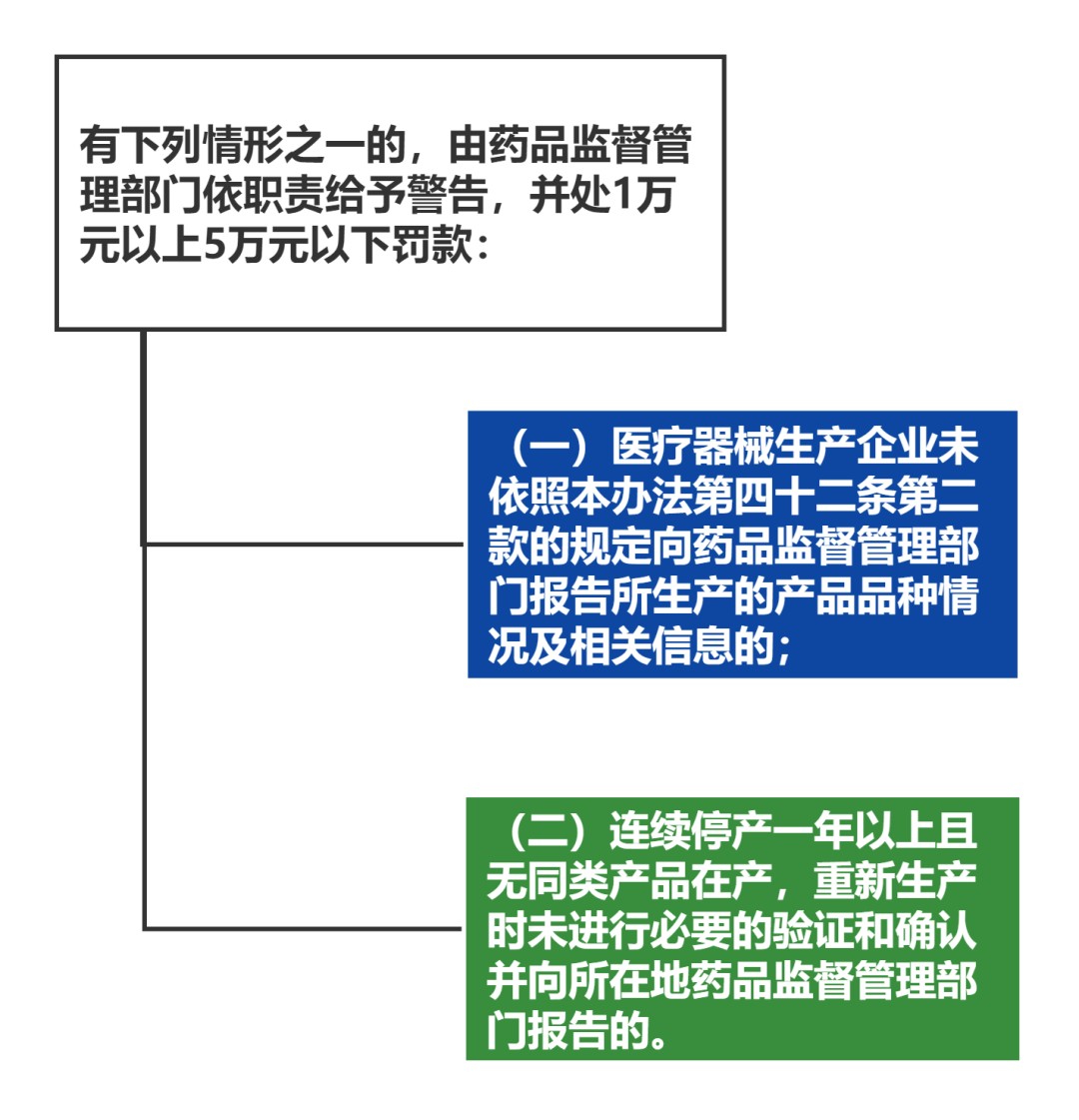
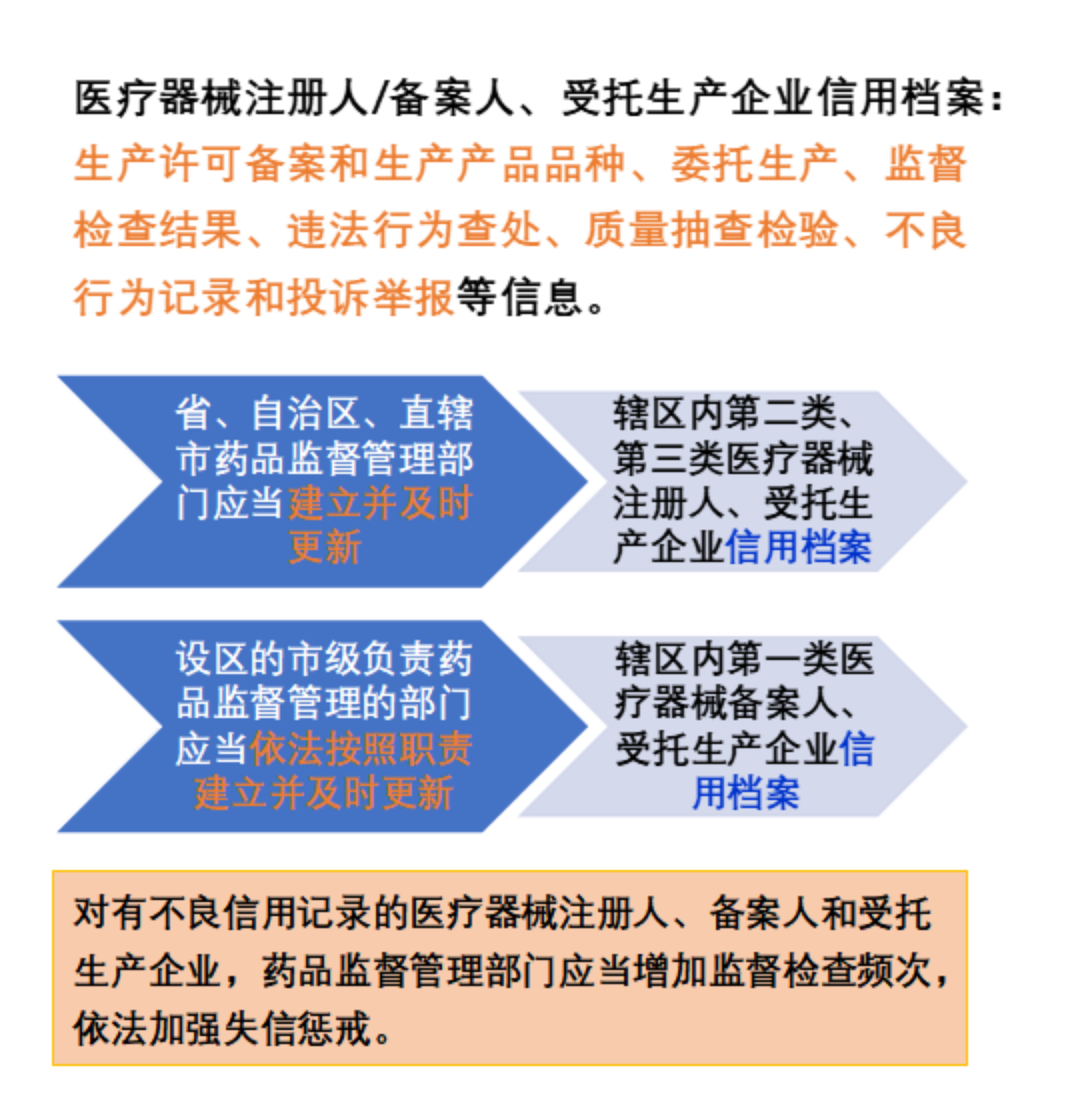
医疗器械生产企业应当向药品监督管理部门报告所生产的产品品种情况。
医疗器械注册人、备案人、受托生产企业应当每年对质量管理体系的运行情况进行自查,并于次年3月31日前向所在地药品监督管理部门提交自查报告。进口医疗器械注册人、备案人由其代理人向代理人所在地省、自治区、直辖市药品监督管理部门提交自查报告。
监督检查

生产的医疗器械对人体造成伤害或者有证据证明可能危害人体健康的,药品监督管理部门可以采取暂停生产、进口、经营、使用的紧急控制措施,并发布安全警示信息。
法律责任
医疗器械生产的违法行为,医疗器械监督管理条例等法律法规已有规定的,依照其规定。