原创
摘要:MDCG 2023-7明确了植入类器械和III类器械豁免临床研究的要求,以及器械等同性证明数据获取层级表
合集:#欧盟MDR法规合集
近日,MDCG发布了一则重磅指南 MDCG 2023-7,阐明了根据MDR Article 61(4)-(6)豁免实施临床研究的要求,以及等同性证明所需数据获取的充分水平。
植入类器械及III类器械豁免临床研究的要求
根据MDR Article 61(4)要求对植入类器械和III类器械进行临床研究,除以下四种情况:

以下四张流程图是对植入类器械和III类器械可否免于强制性临床研究的判定。
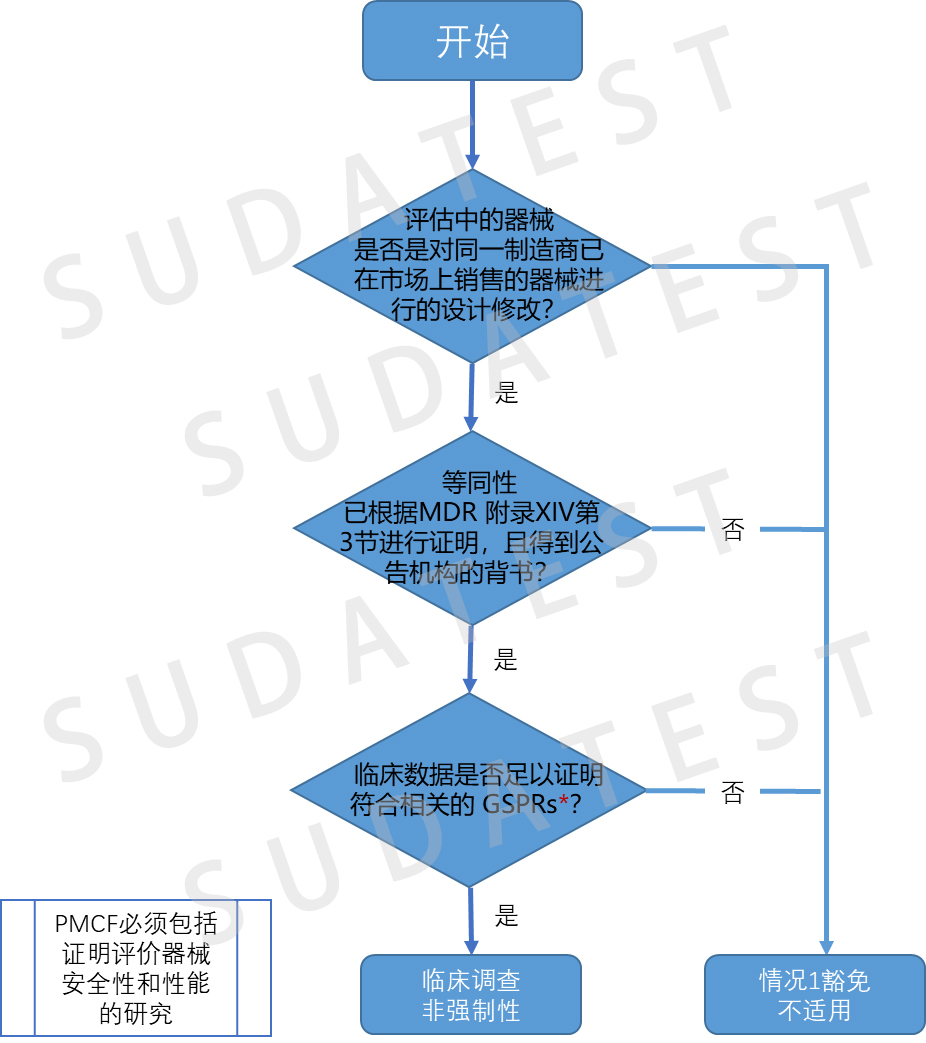
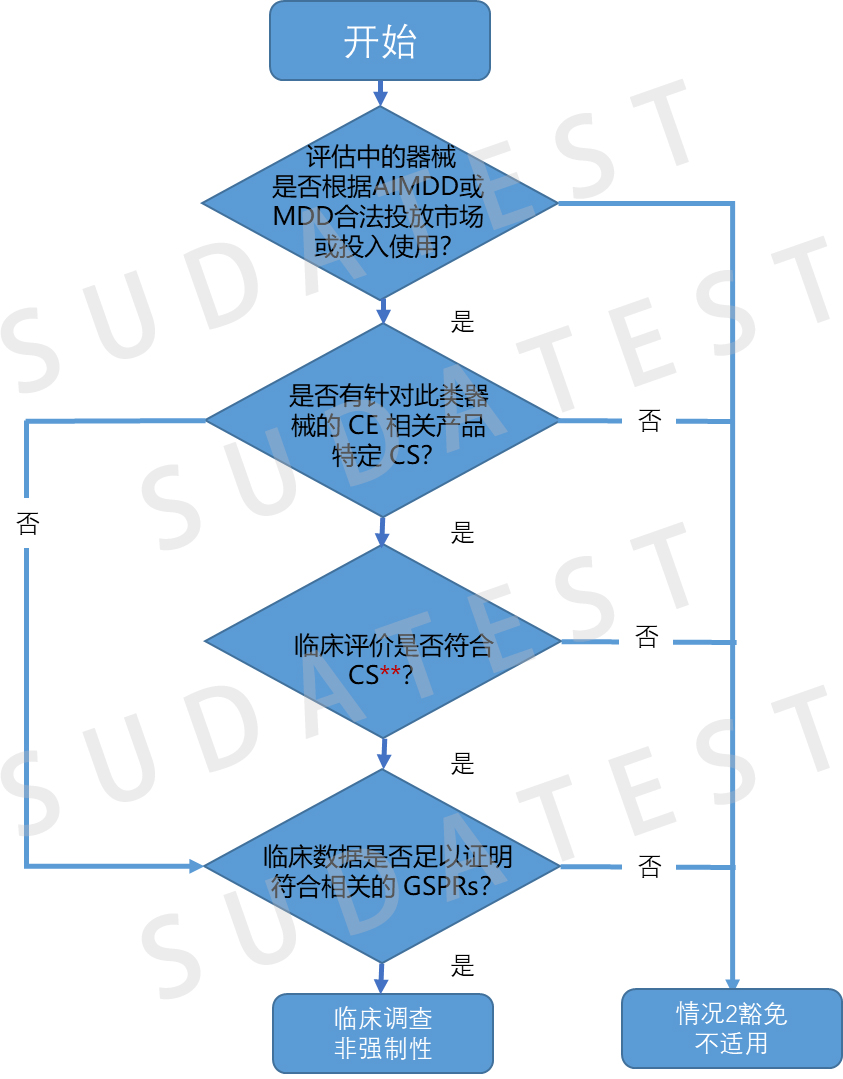
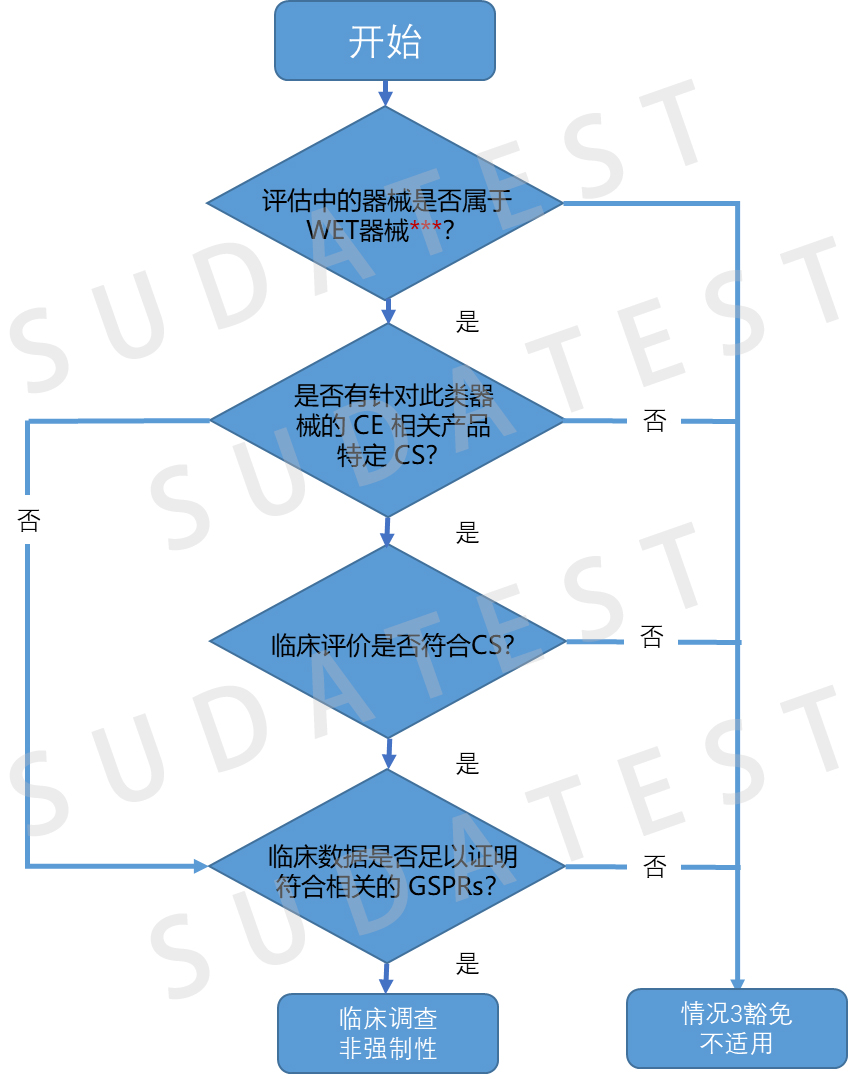
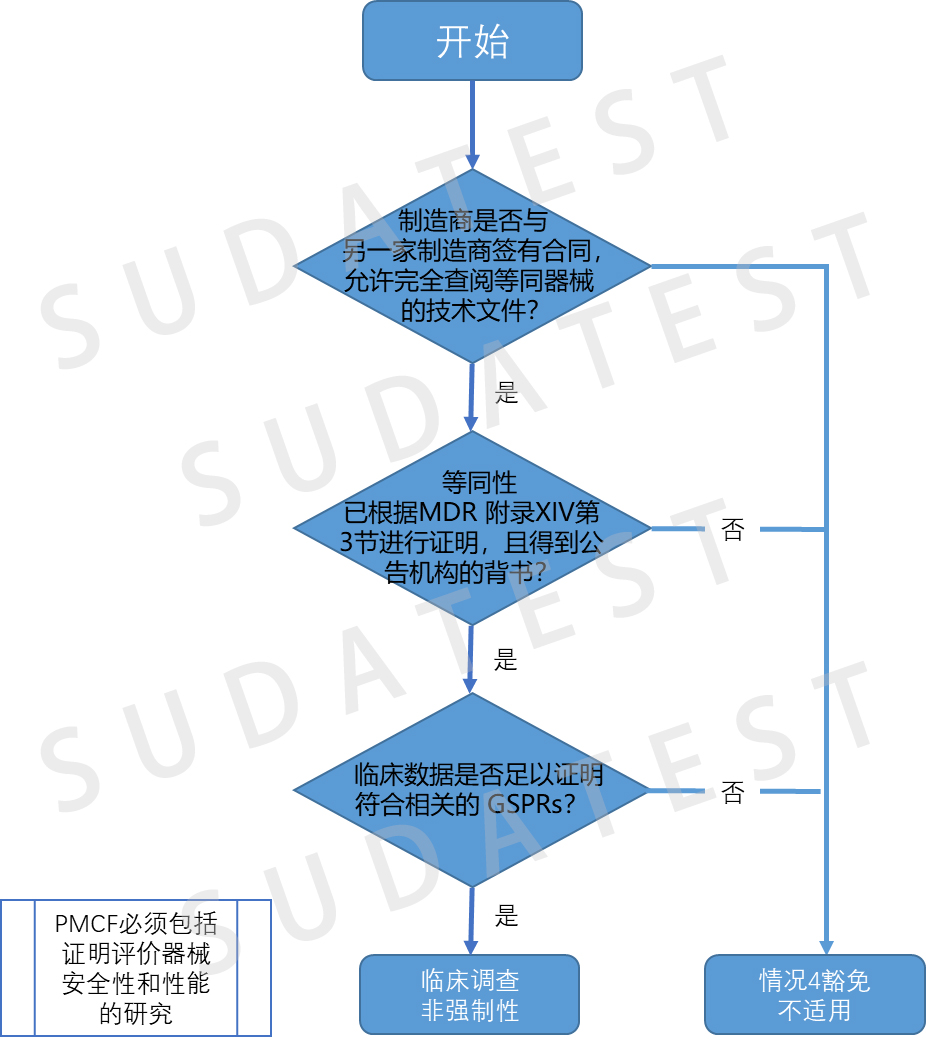
值得注意的是,这四个免于临床研究的情况相互独立,这表明,除非直接引用,否则其中一种情况中概述的条件不适用于其他情况。
在满足MDR 附录XIV第3节描述的等同性标准的前提下,从情况1到情况3,医疗器械制造商可以在不用签订合同的条件下,在评估中的器械的临床评价中使用另一制造商等同器械生成的临床数据。
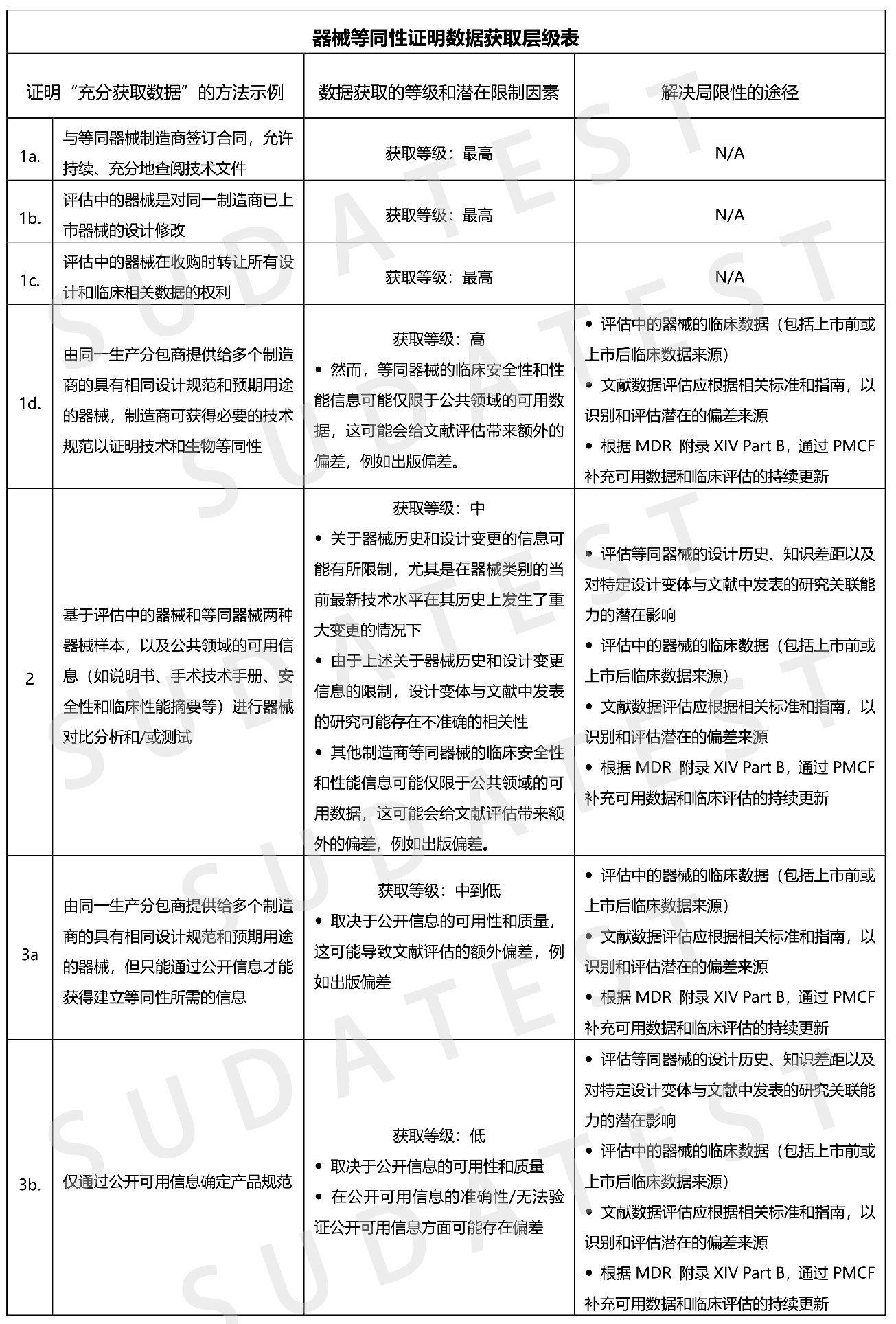
等同性证明所需数据获取的充分水平
除了要从技术、生物和临床三方面考虑等同性证明外,MDR 附录XIV第3节还要求制造商“[…]有充分的条件获取与其声称等同的器械相关的数据,以证明其等同性。如上文所述,证明 "数据获取的充分水平"并不要求在所有情况下都签订合同。只有Article 61(5)所述的豁免情况才需要合同。还应注意的是,MDR附录XIV第3节特别提到证明等同性所需的数据:即要求有足够的获取途径以确定评估等同性所依据的临床、技术和生物特征,而不是获取完整的技术文件。
Article 61 (5)中所述的合同被推定为可提供证明等同性所需的全部数据。然而,在不需要合同的情况下,其他获取数据的手段也足以支持等同性证明,例如上文提到的情况1、2 和3以及情况4的一个特定子集。在该子集中,等同性是通过对多个制造商的多种器械进行充分证明的,并且至少与其中一个制造商签订了合同。评估中的器械制造商必须在临床评估报告中记录说明他们获取数据水平充分的理由,且得到公告机构的认可。下表举例说明了获取等同性证明相关数据的方法,并提出获取这些数据的层级。此外,还指出了这些方法的局限性以及解决这些局限性的途径。但下表仅用于说明目的,并非涵盖所有或规定性的要求。
注:
* GSPRs: General Safety and Performance Requirements,通用安全与性能要求。每个在欧盟销售其产品的医疗器械制造商都必须遵守的要求,旨在确保医疗器械的安全性和有效性。
** CS:Common Specification, 通用规范,为器械、生产或系统的法规符合性提供方法。
*** WET:Well-established Technologies,根据MDCG 2020-6, WET指的是稳定的设计,没有大的改动,在所周知安全有效,有很久的上市历史,例如手术缝合线。
参考资料:MDCG 2023-7 Practical application of Articla 61(4)-(6) sufficient level of access (europa.eu)
【供 稿】苏大检测医疗器械事业部法规中心






