2024年8月28日,FDA发布了修订后的《Voluntary Malfunction Summary Reporting(VMSR)Program for Manufacturers》,即制造商自愿故障摘要报告计划最终指南,旨在简化器械故障报告程序。此次修订对原指南进行了小的技术性的修改,以便与最新版本的FDA 3500A表及当前的不良事件代码保持一致。
VMSR背景
FDA上市后不良事件监管主要通过Medical Device Reporting Program进行。当医疗器械制造商意识到与/疑似与医疗器械有关的死亡、严重伤害或故障时,应及时通过“30天报告”对每起事件进行单独上报。FDA每年收到200多万份的MDRs中,故障报告占大多数。为简化报告流程、减轻制造商的负担,同时使FDA和公众更容易识别故障趋势,FDA推出VMSR计划,允许制造商每季度就与某些产品代码有关器械的某些故障提交故障摘要报告,而非单独的“30天报告”。
产品代码符合性申请
VMSR计划主要面向I类和II类医疗器械,尤其是那些由于频繁故障而产生大量报告的器械。想参与VMSR计划的制造商可通过符合条件产品代码清单确认其器械是否符合条件。若器械所属产品代码不在上述清单内,则制造商可提出产品代码符合VMSR计划的申请。制造商可通过 MDRPolicy@fda.hhs.gov向FDA提起申请,并提交以下信息:
公司名称、地址、注册号;
联系人姓名、电话号码、邮件;
完整的器械识别和描述,包括产品代码和审查小组;
申请的完整陈述和理由,包括讨论与产品代码的受益-风险概况和上市后安全性有关的已知信息,以及为什么没有必要进行个别故障报告的理由;
作为申请理由的一部分,制造商应提供FDA先前就器械资格状态发出的任何信函的副本(包括对文件ID#的引用),并说明为在信函中指出的任何问题而采取的任何行动。
不适用于VMSR计划的情况
应报告的死亡和严重伤害;
根据21 CFR 803.53规定的应报告的故障与公共安全事件有关;
应报告的故障是某些器械召回的主题;
器械出现新型应报告事件;
根据21 CFR 803.12(a)提交的补充信息;
进口商和设备用户设施,因为21 CFR Part 803没有要求这两个实体向FDA报告故障。
故障摘要报告说明
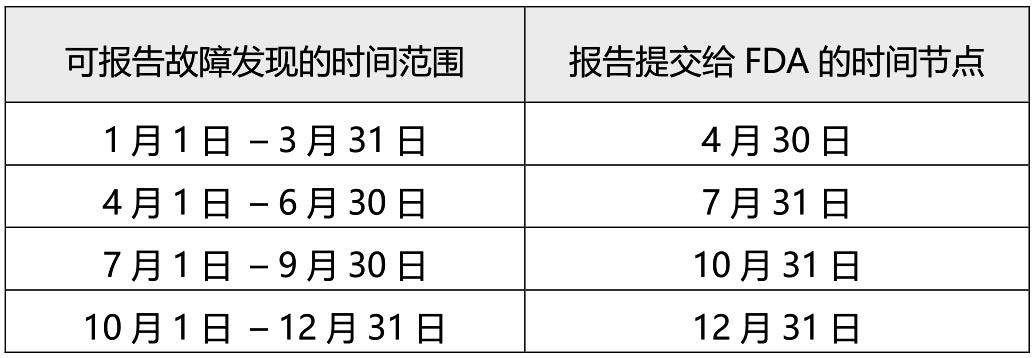
选择参与 VMSR 计划的合格产品代码中的器械制造商必须使用 FDA 3500A 表的适用部分提交故障摘要报告,该报告必须以电子方式提交。故障摘要报告内容包括:器械信息、制造商信息及故障相关信息,故障摘要报告上报时间遵循下表:
指南的末尾还给出了故障摘要报告的示例,有需要的读者可戳下方参考资料。
由于VMSR计划的自愿性,参与的制造商可随时选择退出该计划。退出VMSR计划后,制造商必须重新提交符合要求的“30天故障报告”,并根据需要更新其MDR程序。
参考资料:Voluntary Malfunction Summary Reporting (VMSR) Program for Manufacturers (fda.gov)
【供 稿】苏大检测医疗器械事业部法规中心






