《医疗器械生产质量管理规范附录无菌医疗器械》(2015年第101号)要求应当按照医疗器械相关行业标准要求对洁净室(区)的尘粒、浮游菌或沉降菌等进行定期检(监)测。微生物监测是保证洁净实验室洁净度达标的重要指标,其内容包括非生物活性的空气悬浮粒子数及有生物活性的微生物监测。
研究表明,单一微生物本身并不能独立存在于空气中,他们要么以菌团形式存在,要么必须附着于无活性的尘埃粒子(尤其是直径大于0.5 μm的粒子)之上。因此洁净室空气中的悬浮粒子数量和分布情况与洁净室的洁净度有很大的关系,需有效的控制和准确地监测洁净室中悬浮粒子的数量及其分布,实时掌握洁净室的洁净程度,才能有效的控制空气中的微生物污染。
目前我国关于悬浮粒子监测方法为: GB/T 16292-2010 医药工业洁净室( 区) 悬浮粒子的测试方法,下面将对该方法进行介绍。
相关术语:
洁净室(区):对尘粒及微生物污染规定需进行环境控制的房间或区域。其建筑结构,装备及其使用均具有减少对该区域内污染源的介入、产生和滞留的功能。其他相关参数诸如:温度,湿度、压力也有必要控制。
悬浮粒子:用于空气洁净度分级的空气悬浮粒子尺寸范围在0.1 μm~1 000 μm 的固体和液体粒子。对于悬浮粒子计数测量仪,一个微粒球的面积或体积产生一个响应值,不同的响应值等价于不同的微粒直径。
空态:洁净室(区)在净化空气调节系统已安装完毕且功能完备的情况下,但是没有生产设备、原材料或人员的状态。
静态a:洁净室(区)在净化空气调节系统已安装完毕且功能完备的情况下,生产工艺设备已安装、洁净室(区)内没有生产人员的状态。
静态b:洁净室(区)再生产操作全部结束,生产操作人员撤离现场并经过20min自净后。
动态:洁净室(区)已处于正常生产状态,设备在指定的方式下进行,并且有指定的人员按照规范操作。
测试方法:
本方法采用计数浓度法,即通过测试洁净环境内单位体积空气中含大于或等于某粒径的悬浮粒子数,来评定洁净室(区)的悬浮粒子洁净度级别。
仪器及原理:
测试条件:
在测试之前,要对洁净室(区)相关参数进行预先测试,这类测试将会提供测试悬浮粒子的环境条件,这种预先测试或可包括:
a) 温度和相对湿度的测试。洁净室(区)的温度和相对湿度应与其生产及工艺要求相适应(无特殊要求时,温度在18 ℃~26 ℃,相对湿度在45%~65%为宜),同时应满足测试仪器的使用范围;
b) 室内送风量或风速的测试,或压差的测试;
c) 高效过滤器的泄漏测试。
测试状态
测试报告中应标明测试时所采用的状态和室内的测试人员数。
测试时间
采样点数目及其布置
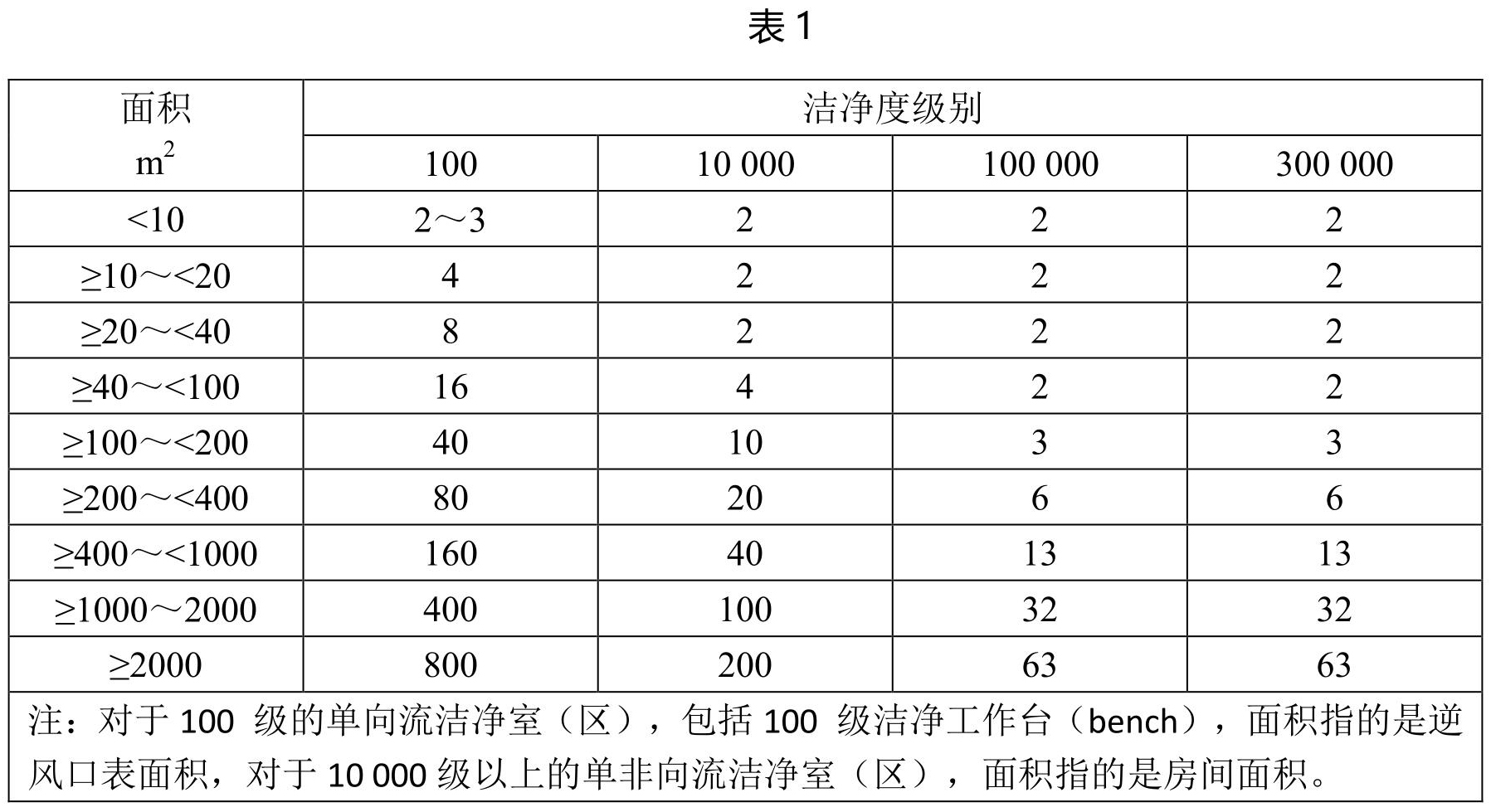
在空态或静态测试时,悬浮粒子采样点数目及其布置应力求均匀,并不得少于最少采样点数目,采样点布置规则见表1。在动态测试时,悬浮粒子采样点数目及其布置应根据产品的生产及工艺关键操作区设置。
采样点的位置:
采样点的位置应满足以下要求∶
a) 采样点一般在离地面0.8 m高度的水平面上均匀布置。
b)采样点多于5点时,也可以在离地面0.8 m~1.5m高度的区域内分层布置,但每层不少于5点。
采用次数的限定:
对任何小洁净室(区)或局部空气净化区域,采样点的数目不得少于 2个,总采样次数不得少于5次。每个采样点的采样次数可以多于1次,且不同采样点的采样次数可以不同。
采样量:
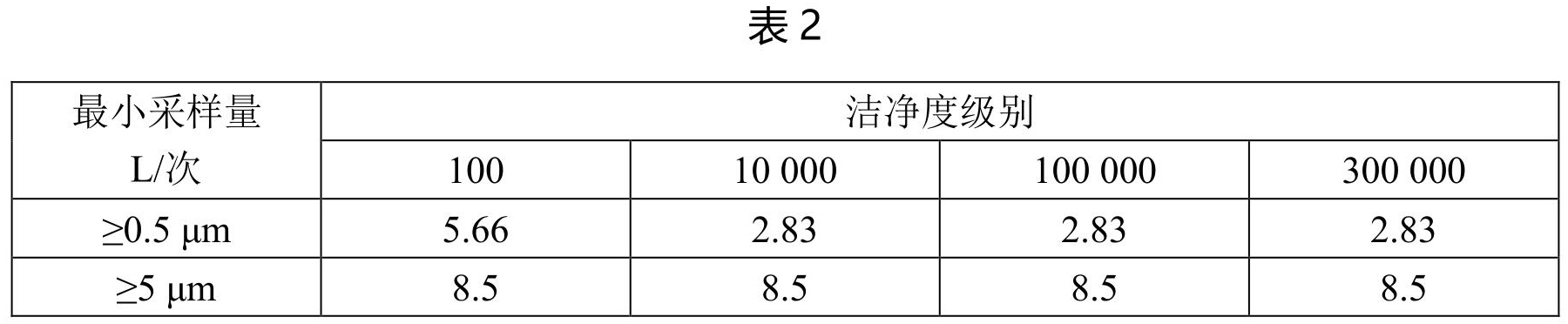
不同洁净度级别每次最小的采样量见表 2。
采样注意事项:
应采取一切措施防止采样过程的污染。
结果计算:
计算每个采样点的平均悬浮粒子浓度(粒/m3)
计算洁净室的平均悬浮粒子浓度(粒/m3)
计算标准差SE。
结果评定:
判断悬浮粒子的洁净度级别应同时满足以下两个条件∶
GB /T16292 - 2010 与 ISO 14644-1: 2015 监测方法比较:
GB /T16292 - 2010 与 ISO 14644-1: 2015 最少采样点数量区别:
【参考资料】GB/T 16292-2010; 《医疗器械生产质量管理规范附录无菌医疗器械》(2015年第101号);国内外药品洁净室悬浮粒子采样点选择及测定结果比对分析[J].中国药品标准,2021,22(06):536-540.
【声明】如需转载须在文首标明来源,且清晰可见!