【声明】版权所有,未经许可不得转载使用!

2022年2月23日,FDA发布了医疗器械质量体系法规(21 CFR Part 820)拟议规则,旨在协调和现行国际医疗器械质量管理体系标准(ISO 13485:2016)的一致性,从而减轻制造商的合规和记录负担。修订后,QSR 820将被修改为QMSR。点击链接查看原文:
QSR 820的由来及修订背景
美国是最先实施医疗器械生产质量管理规范(good manufacturing practice, GMP)的国家之一。早在1987年,美国就施行了《现行药品生产质量管理规范》(cGMP)。1996年,美国对质量管理体系单独立法,公布了《质量管理体系法规》(21 CFR Part 820),简称QSR 820,作为美国医疗器械强制执行的要求。QSR 820删除了先前医疗器械GMP不符合国际规则的部分,使得该规范与ISO 9000的标准更加一致。2012年,美国FDA正式发布指南性文件《医疗器械ISO 13485:2003自愿性审核报告提交程序》(Medical Device ISO 13485: 2003 Voluntary Audit Report Submission Pi-lot Program),允许医疗器械制造商自愿提交认可的第三方认证机构的ISO 13485审核报告,并结合对医疗器械风险管理和评价的结果,降低FDA对医疗器械制造商现场检查的频度。
ISO 13485与QSR 820主要差异(限于篇幅,浅显列示)
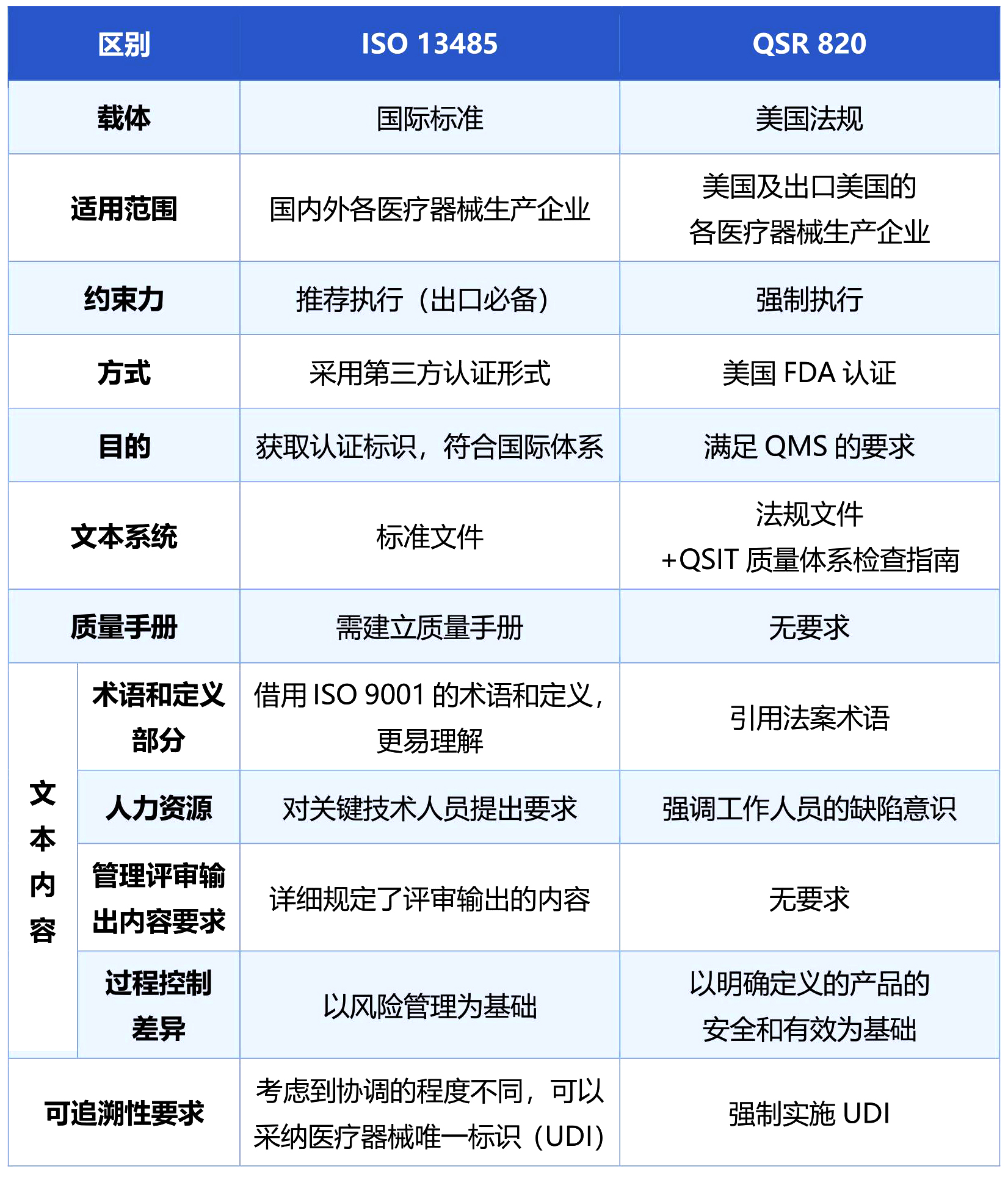
ISO13485现已被大多数国家或地区直接采用或等同采用为本国医疗器械行业的质量管理规范。FDA本次修订目的是将QSR 820与国际公认的质量管理体系监管要求ISO 13485趋同。这意味着国际标准的协调统一发展是未来的趋势,对国内的制造商来说是一个好消息。
参考文献:
[1]陈静,王娟,葛林林,杨晨曦.医疗器械GMP国际化探讨[J].中国医药导刊,2021,23(07):555-560.